1999
2000 2001
2002 2003
2004 2005
2006 2007
2008
2009
geförderte Projekte 1999
Projekt
1999/1
Immuntherapie mit Lymphokin-aktivierten Killerzellen nach allogenen
Stammzelltransplantationen von pädiatrischen Patienten mit akuter
lymphatischer oder myeloischer Leukämie
PD Dr. D. Schwabe, Univ.-Klinikum Frankfurt, Kinderheilkunde
Natürliche Killerzellen sind Immunzellen, die für die Abwehr von Infeken
und von Tumorzellen eingesetzt werden können, ohne dabei den Empfänger
mit schweren Abstoßungsreaktionen zu belasten. Viele Aspekte dieser
experimentellen Therapie im Rahmen einer Übertragung von Stammzellen von
gesunden Eltern auf ihre leukämiekranken Kinder müssen noch erarbeitet
werden.
Mit der Förderung wird ein Spezielmikroskop finanziert, mit dem die
knappen personellen Ressourcen eines laufenden Projektes effektiver
eingesetzt werden können.
nach oben
zur Projektseite
home
In vitro Untersuchungen zu
funktionellen Eigenschaften und Apoptose von T-Lymphozythen in allogenen
Knockenmark- und peripheren Stammzelltransplantaten
Dr. Stefan A. Klein, Univ.-Klinikum Frankfurt am M., Med. Klinik
III
Die Knochenmark- bzw.
Stammzelltransplantation hat in den letzten beiden Jahrzehnten die
größten Fortschritte in der Therapie der akuten Leukämien gebracht. Eine
sehr wichtige, aber dennoch ambivalente Rolle spielen dabei die
T-Lymphzyten (Immunzellen) des Spenders, die sowohl zu Komplikationen
als auch zur Heilung beitragen können. Es handelt sich um die komplexe
Balance verschiedener, z. T. gegensätzlicher Mechanismen.
Dieses Projekt fördert die Untersuchung der Beeinflussung einer
Wirkungs-Balance innerhalb von über-tragenen T-Zellen mitg dem Ziel,
diese besse steuern zu können.
nach oben
zur Projektseite
home
Therapeutische Peptide zur Behandlung der akuten lymphatischen Leukämie (ALL) mit t/9;22)
Dr. Manuel Grez, Georg-Speyer-Haus, Frankfurt
Die Philadelphia-Chromosom-positive akute lymphatische Leukämie ist eine definierte Subgruppe der akuten Leukämie, die mit Chemotherapie allein nicht heilbar ist. Die Entwicklung neuer Therapiekonzepte für diese Erkrankung ist ein Schwerpunkt am Universitätsklinikum Frankfurt am Main.
Das Projekt wird mit 12.000,-- DM unterstützt. Es handelt sich um ein grundlagenorientiertes Projekt, mit dem neue Ansatzpunkte für eine molekulare Therapie untersucht werden sollen. Die Förderung ist als Anschubförderung gedacht, mit der Voruntersuchungen für einen umfangreicheren Antrag bei der Deutschen Forschungsgemeinschaft oder der Krebshilfe ermöglicht werden sollen.
nach oben zur Projektseite homegeförderte Projekte 2000
Projekt 2000/1
Untersuchung zu Apoptose und Alloreaktivität von T-Zellen in allogenen Stammzelltransplantationen, In-vivo-Nachweis und Bedeutung von Leukämiestamm-zellen in autologen peripheren Blutstammzelltransplantaten von Patienten mit akuter myeloischer Leukämie (AML)
Dr. Stefan A. Klein, Med. Klinik III der Joh. Wolfgang Goethe-Universität Frankfurt a. M.
Die allogene Transplantation von blutbildenden Stammzellen aus
Knochenmark (Knochenmarktrans-plantation) oder peripherem Blut
(periphere Blutstammzelltransplantation) ist die Therapie der Wahl zur
Behandlung, ja sogar zur Heilung von Leukämien wie der Chronisch
myeloischen Leukämie (CML) und akuten lymphatischen wie myeloischen
Leukämien (ALL und AML). Der Erfolg der allogenen Transplantation beruht
wesentlich auf der Bekämpfung der Leukämiezellen durch im Transplantat
enthaltene T-Lympho-zyten. Die Bekämpfung der Leukämie durch die Spender
T-Lymphozyten wird als Graft versus Leukemia-Effekt (GvL-, bzw.
Transplantat gegen Leukämie-Effekt) bezeichnet. Über diesen GvL-Effekt
hinaus spielen allogene Spender T-Lymphozyten bei einer Reihe von
weiteren Aspekten der Knochenmarktransplantation eine wichtige Rolle. So
unterstützen sie das Anwachsen der blutbildenden Stammzellen und
verleihen einen Schutz insbesondere gegenüber viralen Infektionen. Die
Kehrseite der Medaille der allogene Spender T-Lymphozyten ist die Graft
versus Host Disease (GvHD, Transplantat gegen Empfänger Erkrankung).
Diese Erkrankung stellt eine Hauptkomplikation nach der allogenen
Transplantation dar. Nahezu zehn Prozent der allogen transplantierten
Patienten versterben an dieser Komplikation. Man geht davon aus, daß der
GvL-Effekt und die GvHD auf den gleichen Mechanismen beruhen. Für den
Erfolg einer allogenen Knochen-marktransplantation ist es von
entscheidender Bedeutung durch das richtige Maß an Unterdrückung der
T-Lymphozyten (d.h. an Immunsuppression) das Risiko der GvHD zu
reduzieren und gleichzeitig einen GvL- Effekt und somit die Bekämpfung
der Leukämie zuzulassen.
Die blutbildenden Zellen zur allogenen Transplantation können durch eine operative Entnahme von Knochenmark oder durch die Sammlung der blutbildenden Zellen aus dem peripheren Blut des Spenders gewonnen werden. Da die Menge dieser Stammzellen im Blut normalerweise sehr gering ist, muß vor der Sammlung der Zellen ihr Anteil im Blut deutlich erhöht werden. Dies geschieht in dem man dem Spender einen körpereigenen Wachstumsfaktor für weiße Blutkörperchen, das G-CSF (Granulozyten-Kolonien stimulierender Faktor) spritzt. Ein Teil der blutbildenden Stammzellen wird durch G-CSF in das Blut ausgeschwemmt und können mit Hilfe eines Gerätes, dem Zellseparator, aus dem Blut gewonnen werden. Die so gewonnenen Transplantate bezeichnet man als periphere Blutstammzelltransplantate (PBSCT).
Transplantate, die mit dieser Methode gewonnen werden, enthalten um ein Vielfaches mehr T-Lymphozyten als konventionelle Knochenmarktransplantate. Trotzdem ist die Rate an Spender gegen Empfänger Erkrankungen (GvHD) bei Patienten die mit peripheren Blutstammzellen transplantiert wurden nicht höher als bei Patienten, die mit Knochenmark transplantiert wurden. In manchen Veröffentlichungen ist sogar von einer niedrigeren GvHD Rate berichtet worden.
Der wesentliche Unterschied zwischen den T-Lymphozyten in den Transplantaten aus Knochenmark und peripherem Blut ist, daß T-Lymphozyten, aus peripherem Blut gesammelt werden, nachdem die Spender mit dem Medikament G-CSF behandelt sind. Die Vermutung, daß dieses Medikament einen Einfluß auf die Funktion der T-Lymphozyten hat, liegt nahe.
Es ist das Ziel dieses Projektes die Ursache bzw. die Mechanismen der veränderten T-Lymphozytenfunktion in peripheren Blutstammzelltransplantaten zu untersuchen. Die Frage nach dem Einfluß des G-CSF auf die T-Lymphozyten in Transplantaten ist kein unwesentliches Detail in der Durchführung der Transplantation. Für die gezielte Steuerung der GvHD und des GvL-Effektes ist sie von großer praktischer Bedeutung.
Es wird diskutiert, daß die Gabe von G-CSF zu einer Veränderung der Produktion von Botenstoffen (Zytokinen) des Immunsystems führt. Dieses veränderte Zytokinmuster hat sicherlich einen Einfluß auf die Funktion der T-Lymphozyten. Anhand von Mäusen konnte früher bereits gezeigt werden, daß bestimmte Botenstoffe des Immunsystems (Interleukin 4 und 10) die Entwicklung einer schweren GvHD hemmen. Gerade diese Zytokine scheinen nach der Gabe von G-CSF vermehrt gebildet zu werden.
Wir konnten mittels der Technik der intrazellulären Zytokinmessung zeigen, daß T-Lymphozyten von Stammzellspendern nach der Gabe von G-CSF in geringerem Maße die Botenstoffe Interleukin 2 und Interferon-gamma bilden. Diese Botenstoffe werden als Th1-Zytokine bezeichnet. Parallel gelang es zu zeigen, daß der Anteil der Zellen die Interleukin 4 (ein Th2-Zytokin) bilden, erhöht ist. Es besteht somit die vermutete Th1Th2 Verschiebung des Zytokinmusters.
Im Rahmen dieser Untersuchungen konnten wir darüber hinaus zeigen, daß
die T-Lymphozyten unter der
G-CSF Gabe sehr leicht sterben. Wir sahen eine deutlich erhöhte Rate an
Zellen, die einer besonderen Form des Zelltodes, der Apoptose bzw. dem
programmierten Zelltod erlagen. Dieser Zelltod trat nach der Aktivierung
der Zellen auf. Wir gehen daher davon aus, daß durch die Veränderung des
Zytokinmusters die Zellen weniger vor der Apoptose geschützt sind. Nach
einer Transplantation würde dies bedeuten, daß T-Lymphozyten bei Kontakt
mit Empfängerzellen, z.B. mit Leukämiezellen sich nicht, wie es sinnvoll
wäre, vermehren, sondern sie sterben und die Leukämie nicht bekämpfen
können.
In weitergehenden Experimenten soll der Mechanismus der veränderten Anfälligkeit der T-Lymphozyten für den programmierten Zelltod untersucht werden. Neben dem Zytokinmuster liegt das Schwergewicht der Untersuchungen auf Veränderungen der Expression von an der Apoptose beteiligten Genen. Ferner wird untersucht welchen Einfluß die G-CSF Gabe auf weitere Funktionen der T-Lymphozyten, wie z.B. dem Erkennen von Krankheitserrregern und von Leukämiezellen hat.
Zusammenfassend tragen diese Untersuchungen bei, den Einfluß der Herkunft bzw. der Gewinnung allogener T-Lymphozyten (Knochenmark oder peripheres Blut) für allogene Stammzelltransplantationen im Hinblick auf den GvL-Effekt, die GvHD und die Wiederherstellung der Funktion des Immunsystems nach der Transplantation zu beschreiben.
siehe auch Bericht 2004nach oben zur Projektseite home
Projekt 2000/2
Translokation (8;21)
bei akuter myeloischer Leukämie (AML) - Etablierung von
in-vivo-Modellen für AML, CML und ALL zur Erprobung
molekulartherapeutischer Ansätze'
Dr. Manuel Grez, Georg-Speyer-Haus Fankfurt a. M.
Leukämien sind Krebserkrankungen des blutbildenden Systems im Knochenmark. Im gesunden Organismus entwickeln sich im Knochenmark aus einer gemeinsamen Vorläuferzelle, der sog. Stammzelle, in mehreren Schritten alle Arten von Blutzellen (rote und weiße Blutkörperchen, Freßzellen, Granulozyten und Blutplättchen). De reifen Zellen treten dann in die Blutbahn über, wo sie spezifische Aufgaben erfüllen. Die Lebensdauer dieser reifen Blutzellen ist allerdings begrenzt, so daß sie im Knochenmark ständig aus Vorläuferzellen nachgebildet werden müssen. Dabei ist es wichtig, daß ein Gleichgewicht zwischen Ausreifung und Neubildung von Blutzellen existiert. Bei Leukämien ist dieses Gleichgewicht gestört, und es kommt zu einer starken Vermehrung von unreifen und funktionsuntüchtigen Vorläuferzellen. Ursachen hierfür sind häufig Schäden (Mutationen) am Erbgut der Vorläuferzellen. Oft handelt es sich dabei um sog. Translokationen, Übertragungen von Chromosomenbereichen zwischen nicht-homologen (unterschiedlichen) Chromosomen.
Gegenstand unseres Projektes ist die Translokation (8;21), die man bei
ca. 40 % der Patienten mit akuter myeloischer Leukämie (AML) vom Typ M2
vorfindet. Bei dieser Translokation kommt es zur Fusion der Gene AML1
und ETO, die normalerweise auf unterschiedlichen Chromosomen lokalisiert
sind. Das so ent-standene Fusionsgen AML1/ETO führt in den betroffenen
Blutzellen dazu, daß diese nicht mehr zu funktionstüchtigen Zellen
ausreifen (differenzieren) können. Ziel unserer Studien ist es, diese
Differenzierngs-blockade spezifisch aufzuheben, was langfristig zur
Entwicklung einer alternativen Therapieform führen sollte. Bislang
werden AML-Patienten einer Chemotherapie, eventuell in Kombination mit
Bestrahlungen, unterzogen. Dabei soll zunächst eine komplette
Krankheitsrückbil-dung (Remission) erzielt werden. Jedoch stellt diese
Behandlungsform eine extreme Belastung für den Patienten dar, da nicht
nur kranke, sondern auch gesunde Zellen abgetötet werden. Außerdem
besteht das Risiko eines Rückfalles (Rezidivs).
Von einer molekularen Therapieform erhoffen wir uns dagegen eine
spezifische Bekämpfung der Leukämie-zellen ohne Beeinträchtigung der
gesunden Zellen, was für den Patienten eine schonendere Behandlungs-form
darstellen sollte.
In der Vergangenheit haben wir zunächst die Mechanismen der durch das
Fusionsgen AML/ETO vermit-telten Differenzierungsblockade untersucht.
Dabei haben wir diejenigen Regionen identifiziert, die für
transkriptionelle Repression, die die Ursache der
Differenzierungsblockade darstellt, verantwortlich sind.
Des Weiteren haben wir herausgefunden, welche Bereiche von AML1/ETO
benötigt werden, um mit anderen Proteinen, die ebenfalls als
transkriptionelle Repressoren bekannt sind, zu interagieren. Die
Ergebnisse dieser Arbeiten wurden Anfang dieses Jahres in einer
anerkannten wissenschaftlichen Zeitschrift ver-öffentlicht (Hildebrand,
D. et al., J. Biol-Chem. 276: 9889 - 98995, 2001). Im Moment
beschäftigen wir uns mit der Entwicklung von Proteinen, die in der Lage
sind, diese Interaktionen spezifisch zu verhindern und so zu einer
Aufhebung der Differenzierungsblockade führen. Wir haben inzwischen ein
Protein konstruiert, das in unserem In-Vitro-Testsystem in der Lage ist,
die durch AML1/ETO vermittelte Repression spezifisch aufzuheben. Zur
Zeit wird die Wirkung dieses Proteins in t(8;21)-Leukämiezellen
untersucht. Darüber hinaus versuchen wir, dieses Protein möglichst so zu
verkleinern, daß es als zellgängiges Peptid in Zellen eingeschleust
werden kann.
geförderte Projekte 2001
Projekt 2001/1
Beihilfe zur Teilnahme zweier Mitarbeiterinnen des Georg-Speyer-Hauses an
einem wissenschaftlichen Kongress 2001
Beide Doktorandinnen arbeiten an der Entwicklung molekularer Therapieformen bzw. der Strukturfunktionsanalyse im Bereich myeloischer Leukämien.
Projekt 2001/2
Therapie der Steroid-refraktären akuten Graft versus Host Disease
Dr. Stefan A. Klein, Med. Klinik III, Joh. Wolfgang Goethe-Universität Frankfurt a. M -Projektstudie-
Die akute Graft
versus Host Disease (akute Transplantat gegen Empfänger Erkrankung,
abgekürzt: aGvHD) ist eine der wesentlichen Komplikationen und die
Haupttodesursache nach der allogenen Knochenmarkstransplantation.
Da immer mehr Fremdspender, und nicht in ihren
Transplantations-antigenen komplett passende Spender (bis hin zur
haploidenten, d. h. nur halb passenden Trans-plantation) für
Knochenmarkstransplantationen herangezogen werden, und außerdem
zunehmend
mehr ältere Patienten transplantiert werden, ist die Häufigkeit der
akuten GvHD zunehmend.
Insbesondere bei einem Befall des Darmes verläuft die akute GvHD bei sehr vielen Patienten tödlich.
Spricht eine initiale Therapie mit hochdosiertem Kortison nicht an, stirbt der Patient mit hoher Wahr-scheinlichkeit. Für Patienten ohne Ansprechen auf Kortison - man nennt eine solche GvHD Steroid refraktär - existiert keine etablierte Therapie.
An der Uniklinik Frankfurt konnten wir bei Patienten mit Steroid refraktärer GvHD sehr gute Erfahrungen mit der Substanz Pentostatin sammeln. Pentostatin ist ein Chemotherapeutikum, das zur Behandlung bestimmter Formen des Lymphdrüsenkrebses eingesetzt wird. Die Substanz ist in der Lage, relativ selektiv die Zellen (T-Lymphozyten), die für die Entstehung der GvHD verantwortlich sind, zu eliminieren.
Aufgrund der guten Therapieergebnisse in Frankfurt haben wir eine multizentrische randomisierte prospektive Studie zur Prüfung von Pentostatin als Therapeutikum der akuten Steroid refraktären GvHD initiiert. Neben der Prüfung von Pentostatin sind weitere Ziele der Studie die Standardisierung des diagnostischen wie auch des therapeutischen Vorgehens bei akuter GvHD.
Zur Diagnostik der GvHD des Darmes setzten wir in Frankfurt seit drei Jahren das schonende und den Patienten wenig belastende Verfahren des Ultraschalls ein. Im Rahmen unserer Studie möchten wir die Wertigkeit dieser Untersuchungstechnik an einer größeren Patientenzahl testen. Ein weiterer wichtiger Aspekt der Studie ist die zentrale histologische (feingewebliche) Untersuchung von Darm-Gewebeproben. Für die histologische Diagnostik, die das wichtigste Verfahren zur Diagnose der akuten Darm-GvHD darstellt, konnten wir dankenswerter Weise Prof. Stolte aus Bayreuth gewinnen, den sicherlich auf diesem Gebiet renommiertesten Pathologen in Deutschland.
An der Studie nehmen mehrere bedeutende Transplantationszentren, wie Wiesbaden, Heidelberg, Frankfurt, Nürnberg, Münster und Dresden teil. Die Studie ist aktiviert und der erste Patient ist bereits eingeschlossen.
Zur Verbreitung der Technik des Ultraschalls des Darmes wurde in Frankfurt im Mai 2002 ein Workshop zur Ultraschall-untersuchung bei akuter Darm-GvHD durchgeführt. Teilgenommen haben Kollegen aus Bad Mergentheim, Essen, Dresden, Hamburg, Heidelberg, Frankfurt, Nürnberg und Münster.
Die Durchführung der Studie zur Therapie der akuten GvHD, insbesondere die zentrale histologische Diagnostik bei Prof. Stolte, wie auch die Durchführung der Veranstaltungen, wie dem Workshop zur Darm-GvHD ist uns nur Dank der Unterstützung durch die Gutermuth-Stiftung möglich.
Zwar ist die GvHD eine häufige Komplikation nach der allogenen Knochenmarkstransplantation, jedoch in absoluten Zahlen eine seltene Erkrankung. Die pharmazeutische Industrie hat daher keinerlei Interesse an der Entwicklung von Therapieverfahren für dieses häufig tödliche Leiden.
Für den wissenschaftlichen Fortschritt in der Behandlung der akuten GvHD und somit für eine Verbesserung der Überlebenswahrscheinlichkeit knochenmarkstransplantierter Patienten ist die Förderung durch ambitionierte Stiftungen, wie der Gutermuth-Stiftung absolut unverzichtbar.
siehe auch:
Bericht über die Therapie der Steroid refraktären akuten Graft versus
Host Disease 2004.
Projekt 2001/3
Bedeutung kleiner GTPasen der Rho-Familie für
die Entstehung Bcr/Abl-positiver Leukämien in vivo
Dr. Gesine Bug, Med. Klinik III, Joh. Wolfgang Goethe-Universität Frankfurt a. M.
Am Beispiel von Ber/Abl positiven Leukämien konnte erstmals die Wirkung
einer tumorspezifischen, molekularen Therapie gezeigt werden. Kleine
GTPasen können die Entstehung von Bcr/Abl positiven Leukämien
beeinflussen, indem sie wichtige Signalwege in der Leukämiezelle ein-
oder ausschalten.
Diese Signalwege zu definieren ist Aufgabe des vorliegenden Projektes.
Durch Verwendung geeigneter Techniken des Gentransfers werden kleine GTPasen in Leukämiezellen gehemmt und die Erbsubstanz mit der in unveränderten Leukämiezellen verglichen. Aus der Analyse von über 6.000 Genen sollen sich neue Ansatzpunkte für eine molekulare Therapie für eine Krankheit ergeben, die durch herkömmlíche Chemotherapien nicht geheilt werden kann.
Die Förderung ermöglicht als Anschubfinanzierung die Durchführung der Genanalyse
nach oben
zur Projektseite
home
Projekt 2001/4
Adaptive Gentherapie für pädiatrische Leukämie durch gentransduzierte
allogene T-Zellen nach Stammzelltransplantation
Projekt der Universitäts-Kinderklinik, Dr. med. Ulrike Köhl, und Georg-Speyer-Haus Ffm., Dr. Manuel Grez
Die jüngsten Fortschritte in der Therapie von akuten Leukämien basieren zu einem nicht unerheb-lichen Teil auf der Weiterentwicklung der allogenen Stammzelltransplantation. Zum einen können durch dosisreduzierte Konditionierungen auch ältere Patienten transplantiert werden, zum anderen werden vermehrt Fremdspen-dertransplantationen oder bei Kindern auch haploidente Transplan-tationen mit Eltern-Stammzellen durch-geführt.
In jedem Fall spielen Graft-versus-Host-Reaktionen eine ganz wesentliche Rolle für den Ausgang einer Transplantation. Je nach Konstellation können chemotherapieresistente Leukämiezellen durch Spender-lymphozyten eliminiert und dadurch Heilungen erzielt werden, oder es entstehen durch eine schwere GvHD lebensbedrohliche Komplikationen. Es ist ein wünschenswertes Ziel, die immunologischen Reaktionen durch Spender T-Zellen besser steuern und individuell den aktuellen Bedürfnissen des Patienten anpassen zu können.
In diesem Projekt wird ein gentherapeutischer Ansatz weiterentwickelt, mit dem sich die Reaktion von Spender-T-Lymphozyten gegen Empfängerzellen gezielt regulieren läßt. Ziel ist es, erwünschte Graft-versus-Leukämie-Effekte induzieren zu können und gleichzeitig in der Lage zu sein, die immunologischen Reaktionen medikamentös zu stoppen, sobald diese in Form einer schweren GvHD außer Kontrolle laufen könnten. Durch gentherapeutische Behandlung (mutante Herpes Simplex Virus Thymidin Kinase HSV-TKS39) soll gleichsam ein "Ausschalter" in die Spender T-Lymphozyten eingebaut werden, der sich durch die einmalige Gabe eines Firusstatikums nach klinischem Bedarf betätigen läßt.
Es handelt sich um ein laufendes Projekt, das in Kooperation mit anderen Zentren durchgeführt wird und aus mehreren Quellen finanziert werden soll. Beantragt wird eine kurzfristige Zwischenfinanzierung.
Zwischenbericht an die Stiftung zum
obigen Projekt
Für viele Kinder mit Leukämie ist die Durchführung einer Hochdosis-Chemotherapie mit anschließender Stammzelltransplantation oft die einzige Möglichkeit die Heilungsrate zu verbessern. Nach Transplantation bietet die Behandlung mit Spender-Lymphozyten (T-Zellen) eine Möglichkeit eine beginnende Abstoßung zu vermeiden, die Zunahme patienteneigener Immunzellen zu revertieren oder zu versuchen, das Risiko eines Rückfalls der Leukämie-Rezidivs zu senken. Bei der Gabe von T-Zellen kann es aber auch zu einer gefährlichen Transplantat-gegen-Wirt-Reaktion (Fachbegriff: graft-versus-host-disease) kommen, bei der sich die T-Zellen gegen gesundes Gewebe des Empfängers richten. Diese graft-versus-host-disease kann verhindert werden durch Ausstattung der T-Zellen mit einem sogenannten Abschaltgen, das nach Aktivierung zur Entfernung der T-Zellen führt.
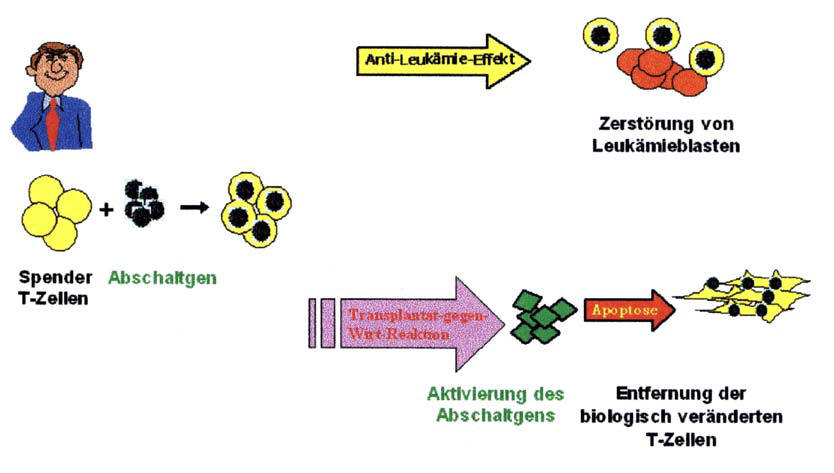
Abbildung 1: Immuntherapie mit biologisch
veränderten T-Zellen
Von einem gesunden Spender werden wichtige Immunzellen (T-Zellen) gewonnen und mit einem Abschaltgen versehen. Diese biologisch veränderten T-Zellen können Leukämiezellen von Patienten effektiv zerstören. Da eine Behandlung mit T-Zellen manchmal auch mit schwerwiegenden Nebenwirkungen verbunden ist, werden diese T- Zellen nach Gabe eines bestimmten Medikamentes (Aktivator des Abschaltgens) zuverlässig entfernt.
In dem vorliegenden Projekt wurde eine neuartige Immuntherapie mit biologisch veränderten Immunzellen zur Behandlung nach Stammzelltransplantation entwickelt. Dazu wurden 2 Vektoren konstruiert, die je ein Abschaltgen und ein sogenanntes Oberflächenmarkergen enthalten. Beide Vektoren konnten nach Untersuchungen an Zelllinien auch erfolgreich ex-vivo in T-Zellen verschiedener Spender eingebracht werden. Es konnte gezeigt werden, dass die biologisch veränderten Immunzellen einen guten anti-leukämischen Effekt gegenüber Leukämiezellen zeigen und zur Zerstörung dieser Leukämiezellen führen. Da diese Immunzellen aber auch Nebenwirkungen verursachen können, wurde durch Inkubation mit bestimmten Medikamenten eine Aktivierung der Abschaltgene eingeleitet. Dies führte zur Elimination der Immunzellen, die besonders effektiv bei der Kombination beider Abschaltgene war. Dies gewährleistet für zukünftige Therapien einerseits eine gute Wirkung gegenüber Leukämiezellen, andererseits aber auch einen guten Schutz vor schweren Nebenwirkungen.
Die Ergebnisse dazu wurden in diesem Jahr publiziert:
Junker K, Koehl U, Zimmermann S, Stein S, Schwabe D, Klingebiel T, Grez M. Kinetics of cell death in T lymphocytes genetically modified with two novel suicide fusion genes. Gene Therapy 14:1189-97 (2003).Für die Anwendung im Rahmen einer klinischen Studie wurde außerdem die Aufreinigung der biologisch veränderten Immunzellen auf ein Verfahren unter „Reinstraumbedingungen“ etabliert
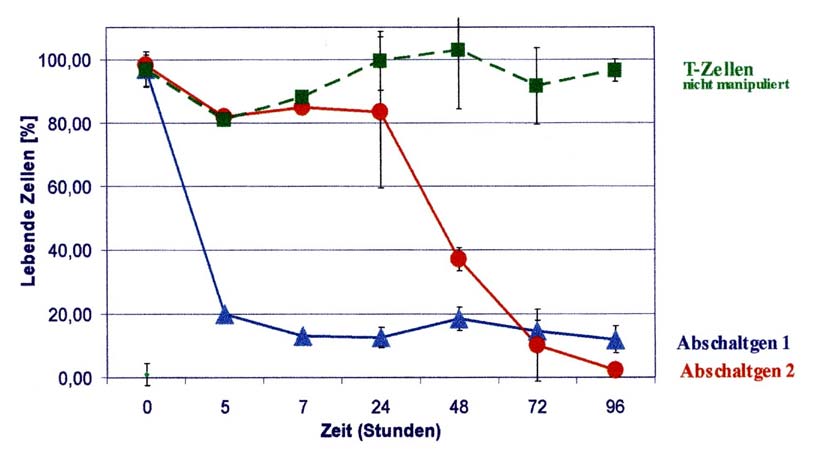
Abbildung 2: Entfernung der T-Zellen durch Abschaltgene
Immunzellen (T-Zellen) eines Spenders, die nicht biologisch verändert sind, bleiben nach Gabe ver-schiedener Medikamente unberührt und lebendig (grüne Linie –quadratisch-). T-Zellen, die ein Abschaltgen enthalten (blaue – dreieckig - und rote Linie – runde -) werden durch Inkubation mit bestimmten Medikamenten nach Aktivierung dieser Abschaltgene effektiv entfernt.
geförderte Projekte 2002
Projekt 2002/1
Risikoprädiktion mittels globaler
Genexpressionsanalyse von Granulozyten bei Patinten mit
myelodysplastischem Syndrom
PD Dr. Wolf-Karsten Hofmann, Med.
Klinik III, Univ.-Klinikum Frankfurt a. M.
D
as myelodysplastische Syndrom (MDS) ist eine Erkrankung der Blutbildung, bei der es zu einem Mangel an roten Blutkörperchen (Blutarmut – Anämie), weißen Blutkörperchen (Abwehrzellen – Granulozyten) und Blutplättchen (Thrombozyten) kommt. Diese Erkrankung beginnt allmählich und kann über viele Jahre unbemerkt verlaufen. Es kommt dann zu klinischen Symptomen (Schwäche, Abgeschlagenheit, gehäufte Infektionen, Blutungen bei Bagatellverletzungen), die den Patienten in der Regel das erste Mal zum Arzt führen. In der Regel sind Patienten jenseits des 60. Lebensjahres von einem MDS betroffen, allerdings erkranken zunehmend junge Patienten, auch Kinder.Eine sehr unangenehme Eigenschaft des MDS ist, dass es die Vorstufe für eine schwere Blutkrebs-erkrankung ist, die akute myeloische Leukämie (AML). Mehr als die Hälfte aller Patienten, die an einem MDS erkranken, entwickeln in einem Zeitraum von 6 Monaten bis zu 15 Jahren eine solche AML. Die AML stellt eine komplizierte Erkrankung der Blutbildung dar, die ohne Behandlung inner-halb von 8 Wochen unweigerlich zum Tode führt. Nur durch intensive Therapiemaßnahmen kann es gelingen, die AML erfolgreich zu behandeln (einschließlich Knochenmarktransplantation).
Die diagnostischen Möglichkeiten beim MDS sind zurzeit noch sehr eingeschränkt. Insbesondere gelingt es mit herkömmlichen Verfahren (mikroskopische Untersuchung, immunologische Unter-suchungen, Analyse der Chromosomen) nicht in allen Fällen, das Risiko der Erkrankung vorherzu-sagen. Seit einigen Jahren gibt es technisch die Möglichkeit, die Regulation und Expression praktisch aller menschlichen Gene in einem Experiment (so genannter DNA-Chip) zu untersuchen.
Wir haben diese Technik hier in Frankfurt seit Januar 2002 etabliert und verfügen bereits über Erfahrungen bei anderen Erkrankungen, insbesondere bei der akuten lymphatischen Leukämie. Mit Hilfe dieser Gesamtanalyse des menschlichen Genoms erhält man ein genaues Abbild der Erkrankung (sogenanntes spezifisches Genexpressionsprofil), mit dem man verschiedene Subgruppen unterscheiden kann.
Ziel des vorliegenden Projektes ist es, solche Subgruppen für das MDS genau zu charakterisieren. Eine rechtzeitige Unterscheidung in Hochrisiko- und Niedrigrisiko-MDS ist erforderlich, um gegebenenfalls frühzeitig eine adäquate Therapie einleiten zu können, die dem Patienten eine Chance auf Heilung gibt.
Projekt 2002/2
Differentielle Genexpressionsanalyse
in hämatopoetischen Stammzellen von Patienten mit myelodyplastischem
Syndrom
PD Dr. Wolf-Karsten Hofmann, Med. Klinik III,
Univ.-Klinikum Frankfurt a. M.
Das myelodysplastische Syndrom (MDS) ist eine Erkrankung der Blutbildung, bei der es zu einem Mangel an roten Blutkörperchen (Blutarmut – Anämie), weißen Blutkörperchen (Abwehr-zellen – Granulozyten) und Blutplättchen (Thrombozyten) kommt. Diese Erkrankung beginnt allmählich und kann über viele Jahre unbemerkt verlaufen. Es kommt dann zu klinischen Symptomen (Schwäche, Abgeschlagenheit, gehäufte Infektionen, Blutungen bei Bagatellver-letzungen), die den Patienten dann das erste Mal zum Arzt führen. In der Regel sind Patienten jenseits des 60. Lebensjahres von einem MDS betroffen, allerdings erkranken zunehmend junge Patienten, auch Kinder.
Eine sehr unangenehme Eigenschaft des MDS ist, daß es die Vorstufe für eine schwere Blut-krebserkrankung ist, die akute myeloische Leukämie (AML). Mehr als die Hälfte aller Patienten, die an einem MDS erkranken, entwickeln in einem Zeitraum von 6 Monaten bis zu 15 Jahren eine solche AML. Die AML stellt eine komplizierte Erkrankung der Blutbildung dar, die ohne Behand-lung innerhalb von 8 Wochen unweigerlich zum Tode führt.
Am Beispiel des MDS konnte bisher durch verschiedene biologische und genetische Methoden gezeigt werden, daß für den Verlauf der Erkrankung von der einfachen Blutbildungsstörung bis hin zur akuten Leukämie die Ansammlung einer Vielzahl von endogenen und exogenen Ereignissen erforderlich ist. Schrittweise wird so die sogenannte Blutstammzelle („Blut-Mutter-zelle“) geschädigt, bis die Schädigung so umfangreich ist, daß diese Zellen bösartig entarten. Auf der Suche nach den Veränderungen in den Stammzellen soll im vorliegenden Projekt die seit einigen Jahren verfügbare Technik der DNA-Chip-Analyse auf diese Blutstammzellen angewandt werden. Damit können bis zu 20.000 menschliche Gene gleichzeitig untersucht werden mit dem Ziel, Defekte zu erkennen und die Zusammenhänge für die Entstehung des MDS zu verfolgen. Wir haben diese Technik hier in Frankfurt seit Januar 2002 etabliert und verfügen bereits über Erfahrungen beim MDS und bei anderen Erkrankungen, insbesondere bei der akuten lymphatischen Leukämie.
Ziel des vorliegenden Projektes ist es, die dem MDS zugrundeliegenden molekularen und genetischen Defekte weiter aufzuklären. Die Anwendung der DNA-Chiptechnik bietet die großartige Möglichkeit, praktisch alle menschlichen Gene in die Untersuchungen einzubeziehen. Das erhöht die Chance beträchtlich, den (oder die) Defekt(e) der Blutstammzelle beim MDS zu finden.
Projekt 2002/3
Mechanismen der Leukämogenese der chimären AML-1 Fusionsproteine
AML-1/ETO und TEL/AML-
PD Dr. Martin Ruthardt, Med. Klinik III, Univ.-Klinikum Frankfurt a. M.
Die Leukämie hat ihre Ursache in der bösartigen Veränderung von einzelnen Zellen im Knochenmark. Je nachdem ob die Leukämiezellen aussehen wie Lymphozyten oder wie Nicht-Lymphozyten spricht man jeweils von lymphatischer Leukämie oder myeloischer Leukämie. Je nach Krankheitsverlauf werden diese beiden Gruppen noch in chronisch und akut unterteilt. Die akuten Leukämien führen unbehandelt innerhalb von Tagen oder weniger Wochen zum Tod, während die chronischen Leukämien einen längeren Verlauf aufweisen. Die Unterscheidung sowohl zwischen chronisch und akut, als auch zwischen lymphatisch und myeloisch ist von großer Bedeutung, da dies für die zu verabreichende Therapie und die Prognose der Patienten entscheidend ist,
Bei über 60%
der Leukämien werden genetische Aberrationen (Abweichungen) für die
Leukämogenese (Entwicklung) verantwortlich gemacht. Die häufigsten
genetischen Aberrationen sind Chromosomentranslokationen, bei denen
Stücke von zwei verschiedenen Chromosomen miteinander verschmelzen.
Damit kommt es auch zur Verschmelzung von zwei Genen die normalerweise
nichts miteinander zu tun haben, zu einem Chimärengen.
Eine Vielzahl dieser Translokationen mit ihrem Chimärengen werden bei
Leukämien gefunden. Sehr häufig ist dabei das Chromosom 21 involviert.
Dabei kommt es zur Fusion des AML-1 Gens mit anderen Genen, wie ETO auf
dem Chromosom 8 bei der Translokation (8;21) oder mit dem Gen TEL auf
Chromosom 12 bei der t(12;21). Die t(8;21) und die t(12;21) gehören zu
den häufigsten Leukämie-assoziierten Translokationen. Die aus diesen
Translokationen abgeleiteten Genprodukte AML-1/ETO und TEL/AML-1 werden
direkt für die Induktion der Leukämie verantwortlich gemacht.
Obwohl beide das AML-1 Gen enthalten führt die t(8;21) mit ihrem Genprodukt AML-1/ETO zur akuten myeloischen Leukämie (AML), während die t(12;21) mit ihrem Genprodukt TEL/AML-1 zur einer Form der akuten lymphatischen Leukämie (ALL), die besonders bei Kindern häufig ist.
Es ist völlig ungeklärt, warum in einem Fall das AML-1 Fusionsprotein zur AML und im anderen Fall zur ALL führt.
Eine Erklärung könnte die Ausgangszelle sein, in
der sich die Translokation ereignet. Somit würde sich die
t(8;21) nur in Zellen ereignen, die myeloisch ausdifferenzieren und die
t(12;21) würde nur in Vorläufern der lymphatischen Zellen stattfinden.
Da Translokationen jedoch zufällige Ereignisse bei der Zellteilung
darstellen, ist diese Hypothese eher unwahrscheinlich. Vielmehr kann man
davon ausgehen, daß sich die Translokationen in sehr frühen noch
pluripotenten hämopoetischen Stammzellen ereignen und daß die Funktionen
der Chimärengene AML-1/ETO und TEL/AML-1 darüber entscheiden, ob sich
eine AML oder eine ALL entwickelt.
Da sich die AML und die ALL sowohl klinisch als auch in ihrem Ansprechen auf die Therapie voneinander unterscheiden, ist es von grundlegender Bedeutung zu verstehen, warum es zur Entwicklung einer AML und nicht zur ALL kommt, und viceversa (umgekehrt). Das könnte die Möglichkeit für neue therapeutische Ansätze für die Therapie der Leukämie eröffnen.
Im hier beantragten Forschungsvorhaben soll untersucht werden, welche Faktoren dazu führen, daß das t(12;21) abgeleitete TEL/AML-1 ALL und das t(8;21)-assoziierte AML-1/ETO AML induziert.
nach oben zur Projektseite home
Differenzieres Monitoring der Immunrekonstitution nach Stammzelltransplan-tation mittels 5-Farb-Flowzytometrie
Klinik für Kinderheilkunde III Pädiatrische Hämatologie, Onkologie und Hämostaseologie der Universitätsklinik Frankfurt - Projektleiter Dr. Ulrike Koehl Leitung des Labors für Stammzelltransplan-
tation und Immuntherapie
Für viele Kinder mit Leukämie ist die Durchführung einer Hochdosis-Chemotherapie mit anschließender Stammzelltransplantation von einem Fremdspender oft die einzige Möglichkeit die Heilungsrate zu verbessern. Nach Transplantation regeneriert sich ein völlig neues Immunsystem aus den transplantierten Stammzellen des Spenders. Die Zeitdauer dieser Zellerholung (medizinischer Begriff: Immunrekonstitution) sowie das Verhältnis der einzelnen Immunzellen zueinander beeinflussen entscheidend die Infektionsabwehr und den Anti-Leukämie-Effekt gegen noch vorhandene Leukämiezellen.
Bisherige
Analysenmethoden zur Bestimmung der verschiedenen Immunzellen nach
Transplantation ermöglichen nur ein ungenaues Bild dieser Zellerholung.
Ziel des vorliegenden Antrages ist es daher, mit Hilfe einer sogenannten
5-Farb-Flowzytometrie ein Analysenverfahren zu entwickeln, um die
unterschied-lichen Immunzellen zu verschiedenen Zeitpunkten nach
Transplantation genau zu bestimmen. Diese neu-artige
5-Farb-Flowzytometrie ermöglicht an einer einzelnen Zelle gleichzeitig
fünf verschiedene Eigen-schaften zu messen. Mit peripherem Blut gesunder
Spender soll zunächst ein Panel mit ver-schiedenen Antikörpern erstellt
werden, um die unterschiedlichen Immunzellen voneinander zu
unter-scheiden. Anschließend soll bei 20 Patienten mit akuter Leukämie
nach erfolgter Stammzelltransplantation die Zellerholung ermittelt
werden. Dazu werden die Analysen der Immunzell-Subpopulationen in den
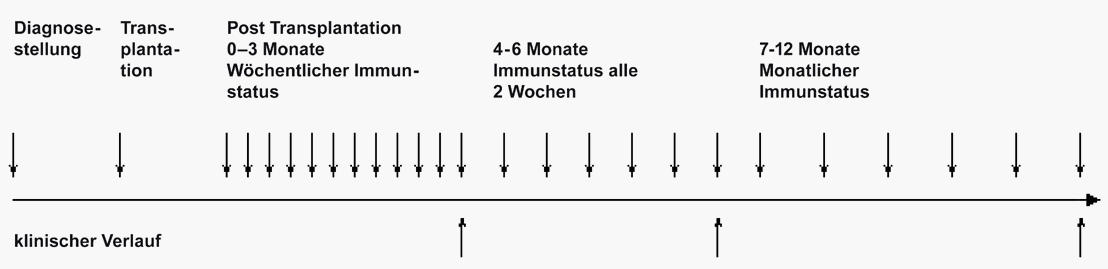
ersten
3 Monaten nach Transplantation wöchentlich, im 4. Monat alle 2 Wochen
und bis 1 Jahr nach Transplantation alle 4 Wochen erhoben.
Kooperationen:
PD Dr. H. Martin, Innere Medizin, Universitätsklinik Frankurt; Prof. Dr.
R. Handgretinger, Director, Division
of Stem Cell Transplantation, St. Jude Children`s Research Hospital, in
Memphis, USA: Entwicklungen von Immuntherapien
Zwischenbericht an die Gutermuth-Stiftung für die Projekte:
1.
Immuntherapie mit Lymphokin-Aktivierten-Killerzellen nach allogener
Stammzelltransplantation von
pädiatrischen Patienten mit akuter lymhpatischer oder
myeloischer Leukämie
2. Monitoring der Immunrekonstitution nach Stammzelltransplantation mittels 5-Farb-Flowzytometrie
Projektleiter
Dr. Ulrike
Koehl Leitung des Labors für Stammzelltransplantation und
Immuntherapie
Klinischer Hintergrund
Für viele
pädiatrische Patienten mit einer malignen Erkrankung ist die
Durchführung einer Hochdosis-Chemotherapie mit anschließender
Stammzell-Transplantation von einem Fremdspender oft die einzige
Möglichkeit, die Heilungsrate zu verbessern. Wird die toxische
Chemotherapie in hohen Dosen verabreicht, so werden die Krebszellen
effektiver getötet. Leider zerstört die Chemotherapie auch alle
Knochenmarks-zellen incl. der Knochenmark-Stammzellen, aus denen alle
Abwehrzellen des Blutes gebildet werden.
Nach Transplantation bildet sich ein neues Immunsystem aus den
Stammzellen des Spenders. Einige Patienten erleiden nach
Stammzelltransplantation einen erneuten Rückfall ihrer Erkrankung oder
versterben an schweren Infektionen. Dies gilt insbesondere für
Patienten, bei denen sich das neue Immunsystem nur sehr langsam erholt.
Es ist daher dringend notwendig, bei diesen Patienten die Zellerholung
nach Transplantation zu beschleunigen.
Zusammenfassung der Ergebnisse aus den laufenden Projekten
Im Rahmen des 1. Projektes wurde im
Labor für Stammzelltransplantation und Immuntherapien ein Ver-fahren
entwickelt, um natürliche Killerzellen (NK-Zellen) des Fremdspenders
unter Reinstraumbedingungen aufzureinigen und von aggressiven T-Zellen
zu befreien (Abb.1, Lit.1). Natürliche Killerzellen des Spenders sind in
der Lage, die Neubildung der Blutzellen zu beschleunigen.
Dies wird derzeit genauer im Rahmen des 2. Projektes untersucht. Darüber
hinaus sind Natürliche Killer-zellen maßgeblich an der Immunabwehr gegen
Tumoren und Leukämien sowie an der Abwehr gegen schwere Infektionen
beteiligt (Lit. 2).
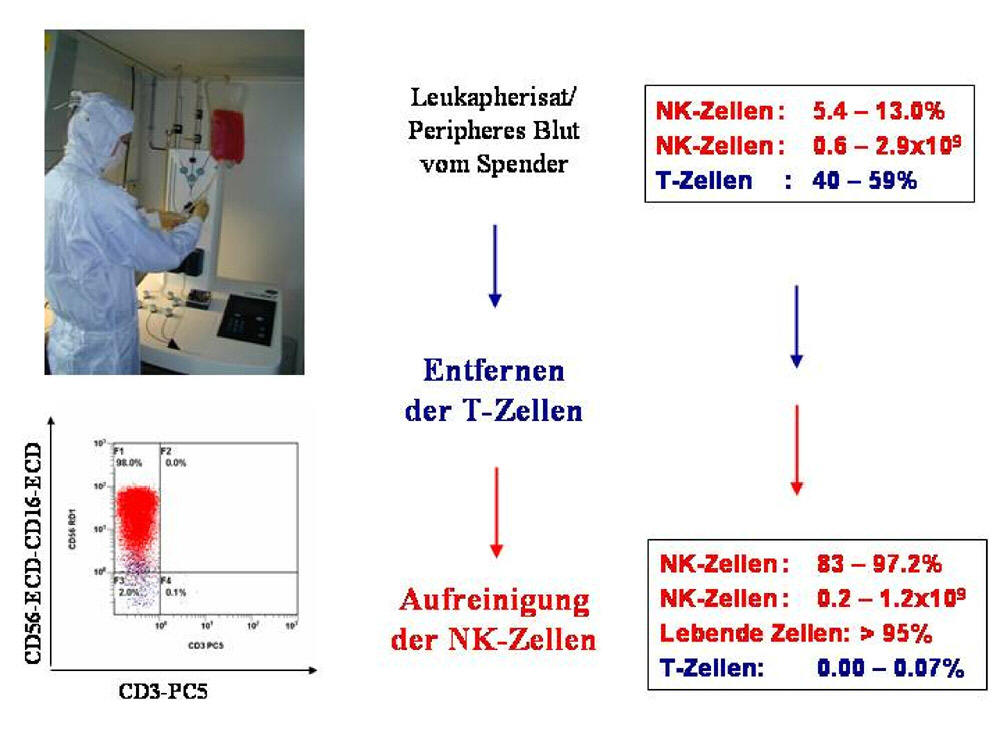
Abbildung 1: Aufreinigung und
Herstellung von NK-Zellpräparaten zur Immuntherapie
Von einem gesunden Spender werden wichtige Immunzellen (Natürliche
Killer-Zellen, abgekürzt NK-Zellen) gewonnen. Dazu wird dem Spender
zuerst peripheres Blut entnommen. Dieses enthält 5-14% der wichtigen
NK-Zellen, aber auch 40-60% T-Zellen, die für den Patienten gefährlich
sind und daher durch aufwendige Selektionsverfahren unter
Reinstraumbedingungen (Bild oben links) entfernt werden müssen.
Zuerst werden die Zellen mit Antikörpern markiert, die nur an die
T-Zellen binden. An diesen Antikörpern hängen magnetische Kügelchen,
wodurch die T-Zellen in einem starken magnetischen Feld von den nicht
markierten Zellen getrennt werden können. Anschließend werden die
restlichen Zellen mit Antikörpern gegen die NK-Zellen markiert, um diese
von allen anderen Zellen zu trennen. Nach der Aufreinigung enthält das
Zell-präparat bis zu 97% NK-Zellen und weniger als 0,07% T-Zellen. Die
Herstellung solch einer Immuntherapie dauert 18-28 h. Als
Qualitätskontrolle werden neben Untersuchungen auf Verunreinigungen mit
Bakterien und Pilzen auch so genannte FACS-Analysen durchgeführt. Damit
können die einzelnen Zellen in einem Präparat genau dargestellt werden
(siehe als Beispiel die längliche rote Punktwolke im linken
unteren Bild, das die aufgereinigten NK-Zellen zeigt).
Bei dieser neuartigen Immuntherapie erhält
der Patient von seinem Spender außer den lebensnotwendigen Stammzellen
am Tag 3, 40 und 100 nach Transplantation zusätzlich NK-Zellen (Abb. 2;
Lit. 3, 4).
In einer ersten Machbarkeitsstudie wurden 3 Kinder mit Leukämien in
einer aussichtslosen Situation be-handelt. Bei allen 3 Patienten konnte
eine Lebensverlängerung erreicht werden (Lit. 3).
Derzeit sind in einer laufenden Studie 5 pädiatrische Patienten in
Frankfurt und 5 internistische Patienten in Basel mit dieser Therapie
behandelt worden. 9 von 10 Patienten geht es gut, alle zeigen eine gute
Lebens-qualität.
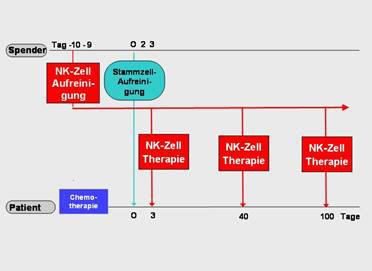
Abbildung 2: Immuntherapie mit
natürlichen Killerzellen nach Stammzelltransplantation
Von einem gesunden Spender
werden wichtige Immunzellen (Natürliche Killer-Zellen, abgekürzt
NK-Zellen) gewonnen, vermehrt und teilweise eingefroren. Nach
Stammzelltransplantation (am Tag 0) erhält der Patient zusätzlich an den
Tagen 3, 40 und 100 eine Immuntherapie mit NK-Zellen.
Literatur
1. Koehl U, Esser R, Zimmermann S, Sörensen J, Grüttner HP, Martin H, Kotchetkov R, Bartling T, Lang P, Tonn T, Seidl C, Seifried E, Klingebiel T, Schwabe D Clinical scale selected and expanded CD56+ CD3- donor cells for immunotherapy. Immonology, Medimond 2004, 407-415.
2. Zimmermann S, Esser R, Klingebiel T, Koehl U. A novel four-colour-flow cytometric assay to determine NK cell or T cell mediated cellular cytotoxicity against leukemic blasts in peripheral blood or bone marrow specimen containing greater than 20% of normal cells. J Immunol Methods 2005, 296: 63-76.
3. Koehl U, Sörensen J, Esser R, Zimmermann S, Grüttner HP, Tonn T, Seidl C, Seifried E, Klingebiel T, Schwabe D. IL-2 activated NK cell immunotherapy in three children after haploidentical stem cell transplantation. Blood Cells, Mol. and Diseases 2004, 33: 261-266
4. Passweg JR, Stern M, Koehl U, Uharek L, Tichelli A. Use of natural killer cells in hematopoetic stem cell transplantation. Bone Marrow Transplantation 2005, 17: 15-21.
Ergebnis
veröffentlicht in:
Abstract
http://www.ncbi.nlm.nih.gov/pubmed/19701252
nach oben zur Projektseite home
Einsatz von mesenchymalen Stammzellen bei Patienten mit akuter Leukämie
Dr. Kathrin Rieger,
Medizinische Klinik III, Charitè - Campus Benjamin Franklin,
Hochschulmedizin Berlin
Trotz zunehmender
Fortschritte in der Therapie der akuten Leukämien ist für die Mehrheit
der Patienten eine dauerhafte Heilung Ihrer Erkrankung weiterhin nur
durch eine Knochenmark- bzw. Stammzelltransplantation möglich.
Leider ist diese Therapieform auch mit ernsten Risiken wie z. B. dem
Transplantatversagen oder der sogenannten
Spender-gegen-Empfänger-Erkrankung, in der sich das neue Immunsystem
gegen die Organe des Transplantat-Empfängers richtet, verbunden.
Die Sicherstellung der Transplantation und die Regulierung des Immunsystems nach einer Transplantation sind daher von großer Bedeutung. Neuere Forschungen konnten zeigen, daß hierbei die sogenannten mesenchymalen Stammzellen (MSC) eine wichtige Rolle spielen.
Mesenchymale
Stammzellen sind Ursprungszellen u. a. für das Bindegewebe. Sie besitzen
eine hohe Regenerationsfähigkeit, können aus dem Knochenmark angezüchtet
werden und sind in der Lage, ganz unterschiedliche Gewebe aufzubauen.
Wir wissen heute, daß diese mesenchymalen Stammzellen sowohl das
Anwachsen des Stammzell-transplantats verbessern, als auch das Auftreten
und den Schweregrad der Spender-gegen-Empfänger-Erkrankung vermindern
können.
Unsere
Arbeitsgruppe befaßt sich mit der Anzüchtung der mesenchymalen
Stammzellen und der Erprobung dieser Zellen als Beimischung zu
herkömmlichen Stammzelltransplantaten bei Patienten mit akuter Leukämie.
Wir konnten zeigen, daß sich mesenchymale Stammzellen mit einem hohen
Reinheitsgrad vermehren und züchten lassen.
Der nächste Schritt unserer Forschung wird sein, diese Zellen klinisch
einzusetzen.
nach oben zur Projektseite home
Projekt 2002/6
Genexpressionsanalyse residualer
leukämischer Blasten bei Kindern mit akuter lymphoblastischer Leukämie
(ALL)
Peter Rhein, Dipl.-Ing./Biotechnologie (Doktorand)/Dr. Leonid Karawajew,
Robert-Rössle-Klinik, Charité, Humboldt Universität Berlin
Die akute lymphoblastische Leukämie
(ALL) ist die häufigste Krebserkrankung im Kindesalter. Sie besitzt
in Deutschland eine Inzidenz von ca. 600 Fällen pro Jahr. Die
Überlebensrate ist abhängig von den initialen Risikofaktoren und beträgt
heute durch den Einsatz von Chemotherapeutika im Durchschnitt 75 %.
Für einen bestmöglichen Verlauf der Chemotherapie ist die Einteilung der
Patienten in Risikogruppen (Stratifizierung) notwendig, nach der sich
das Therapieprotokoll richtet. Das Therapieansprechen ist der für die
Stratifizierung entscheidende Risikofaktor. Um ihn zu bestimmen, wird
die Zahl der leukämischen Restzellen (residualer Blasten) gemessen, die
nach Beginn der Therapie noch vorhanden sind.
Die große klinische Bedeutung der
residualen leukämischen Zellen macht sie für eine molekularbiologische
Charakterisierung wertvoll. Die moderne Genomforschung bietet hierfür
völlig neue Möglichkeiten. So stehen nach der Aufklärung der Struktur
des menschlichen Genoms neue, leistungsfähige Technologien zur
automatisierten Funktionsanalyse der Gene zur Verfügung. Eine solche
Methodik ist die auf so genannten DNA-microarray-Chips basierende
Genexpressionsanalyse von Zellen. Bei dieser Methode sind kurze
Gensequenzen an einen Chip gebunden, durch die nahezu alle Gene der
menschlichen Zelle repräsentiert werden. Anschließend können
Patienten-Proben auf den Chip aufgetragen und gemessen werden, wodurch
bestimmt wird, wie stark jedes einzelne Gen ursprünglich expremiert
worden ist.
Das Ziel des Projektes ist die Isolierung der während der Therapie
persistierenden (verbleibenden) leukämischen Zellen sowie deren
Charakterisierung anhand von Genexpressionsprofilen. Die so gewonnenen
Daten werden mit modernsten bioinformatischen Methoden analysiert und
dienen dazu, Gene, die an der Entstehung von Krankheiten beteiligt sind,
auf molekularer Ebene zu untersuchen. Dies ermöglicht, Ursachen der
Krankheitsentstehung zu erkennen und neue Wirkstoffe und Therapien zu
entwickeln.
nach oben zur Projektseite home
geförderte Projekte 2003
Risikoprädiktion Mittels Globaler Genexpressionsanalyse von Granulozyten bei Patienten mit myelodysplastischem Sandrom
PD Dr. Wolf-Karsten Hofmann, Med. Klinik III, Univ.-Klinikum Frankfurt a. M.
Die diagnostischen Möglichkeiten beim MDS sind zurzeit noch sehr eingeschränkt. Insbesondere gelingt es mit herkömmlichen Verfahren (mikroskopische Untersuchung, immunologische Untersuchungen, Analyse der Chromosomen) nicht in allen Fällen, das Risiko der Erkrankung vor-herzusagen. Seit einigen Jahren gibt es technisch die Möglichkeit, die Regulation und Expression praktisch aller menschlichen Gene in einem Experiment (so genannter DNA-Chip) zu untersuchen.
Diese Technik
wurde in Frankfurt seit Januar 2002 etabliert. Es gibt bereits
Erfahrungen bei
anderen Erkrankungen, insbesondere bei der akuten lymphatischen
Leukämie. Mit Hilfe dieser Gesamtanalyse des menschlichen Genoms erhält
man ein genaues Abbild der Erkrankung (sogenanntes spezifisches
Genexpressionsprofil), mit dem man verschiedene Subgruppen unterscheiden
kann.
Ziel des vorliegenden Projektes ist es, solche Subgruppen für das MDS genau zu charakterisieren. Eine rechtzeitige Unterscheidung in Hochrisiko- und Niedrigrisiko-MDS ist erforderlich, um gegebenenfalls frühzeitig eine adäquate Therapie einleiten zu können, die dem Patienten eine Chance auf Heilung gibt.
nach oben
zur Projektseite
home
PD Dr. Wolf-Karsten Hofmann, Med. Klinik III, Hämatologie, Onkologie, Transfusionsmedizin,
Campus Benjamin Franklin, Charité Univ.-Medizin 12203 Berlin, Hindenburgdamm 30
Anschaffung des 2100 Bioanalyzer Desktop
System, zur Fortsetzung seiner
wissenschl. Forschungsarfbeit
in Berlin
nach oben
zur Projektseite
home
Untersuchung der altersabhängigen Erholung bei Kindern und Erwachsenen
Dr. Ulrike Köhl, Labor für Stammzelltransplantation und Immuntherapie, Abt. für Pädiatrische Hämatologie und Onkologie, Klinikum der J.W. Goethe-Universität Frankfurt a. M.
Die
Knochenmark-Stammzelltransplantation von Fremdspendern ist eine
wirkungsvolle Therapie bei manchen Kindern und Erwachsenen mit Leukämie.
Aus dem transplantierten Knochenmark ent-wickelt sich im Laufe von
Monaten ein neues Immunsystem, dem es gelingt, die Leukämiezellen zu
bekämpfen. Allerdings ist dieses sich neu entwickelnde Immunsystem erst
nach und nach in der Lage, infektiöse Erreger - Bakterien, Pilze und
Viren - schnell und effizient zu bekämpfen.
Im Labor für Stammzelltransplantation und Immuntherapie der Uniklinik
Frankfurt wurde im letzten Jahr dank der Unterstützung der
Gutermuth-Stiftung eine verbesserte Methode zur Messung der
Immunrekonstitution nach Stammzelltransplantation entwickelt. Darauf
aufbauend soll im Verlauf des ersten Jahres nach Transplantation
regelmäßig und engmaschig der Anteil der verschiedenen Immunzellarten
im Blut gemessen werden.
An mehreren Zeitpunkten soll zudem die Reaktivität des Abwehrsystems
gegen Viren, Pilze und Leukämie-zellen getestet werden (Grafik.) Die
gemessene Aktivität soll mit der Zunahme der verschiedenen Zellen des
Immunsystems verglichen werden. Ergänzend soll untersucht werden, ob
besondere genetische Merkmale ("Polymorphismen") mit einer verminderten
Immunabwehr gegen Infektionen oder auch Leukämiezellen zusammenhängt. Im
Anschluss an die Datenerhebung erfolgt eine statistische Auswertung, um
nach Möglichkeit „Hochrisikopatienten“ zu beschreiben. Die Ergebnisse
sollen verwendet werden, die Therapie der nachfolgenden Patienten weiter
zu verbessern
nach oben zur Projektseite home
Inhibition des Wnt-Signalweges durch Inhibitoren der Cycloxygenase-1 als Beitrag zur Therapie der akuten myeloischen Leukämie
Dr. Elena Pucetti, Med. Klinik III, Klinikum der J.W. Goethe-Univ. Frankfurt a. M.
Wir konnten kürzlich beweisen, daß
eine gestörte "Selbsterneuerung" der hämopoetischen Stammzellen eine
bedeutende Rolle für die Leukämogenese spielt, indem wir zeigen konnten,
dass ein bestimmter Signalweg, der Wnt-Signalweg, durch die Expression
von PML/RARa und AML-1/ETO aktiviert wird. Der Wnt-Signalweg setzt
Signale, welche durch die Aktivierung eines biologischen Schalters durch
die Bindung an ein bestimmtes Protein, dem sog. Wnt, auf der
Zelloberfläche initiiert werden, in neue Zellaktivitäten um. Es wurde
kürzlich gezeigt, daß die Aktivierung des Wnt-Signal-wegs die
Selbsterneuerung von normalen hämopoetischen Stammzellen stark erhöht.
Die AML-assoziierten Fusionsproteine aktivieren den Wnt-Signalweg über
die Hochregulierung von
y-catenin, eines Schlüsselfaktors im Wnt-Signalweg. Die Inhibition des
Wnt-Signalweges durch die Unterdrückung der g-catenin-Expression durch
eine neuartige Technik, der sog "RNA interference", hebt
die gestörte "Selbsterneuerung" von PML/RARa- und
AML-1/ETO-exprimierenden hämopoetischen Stammzellen auf. Die Bedeutung
der aberranten Aktivierung des Wnt-Signalwegs für die Leukämogenese wird
auch dadurch bestätigt, daß die Überexpression von g-catenin zu Leukämie
in Mäusen führt.
Der Wnt-Signlweg kann pharmakologisch gehemmt werden. Dabei kommt den
sog nicht steriodalen Entzündungshemmern (NSAID) aus der Gruppe der
Cyclooxagenase-1 Inhibitoren, zu denen so einfache Schmerzmittel wie
Aspirin, Sulindac, Etodolac u.a. gehören, eine große Bedeutung zu. Es
konnte kürzlich gezeigt werden, daß ein Derivat des Aspirins in der Lage
ist, den Wnt-Signalweg zu unterbinden. Diese Substanz wurde als
magenschonende Variante von Aspirin entwickelt, ohne dessen
Hauptwirkungen zu beeinflussen. Außerdem ist bekannt, daß Aspirin eine
gewisse vorbeugende Wirkung gegen Tumoren im Allgemeinen hat.
Ziel des Projektes ist es, die Möglichkeitender NSAIDs in der Therapie
der AML auszuloten. Dazu sollen zuerst an den vorhandenen Zellmodellen
der AML der Einfluß von Sulindac und NO-ASA auf die aberrante
Selbsterneuerung von Stammzellen untersucht werden, welche die
AML-assoziieten Fusionsproteine exprimieren. Anschließend soll in
unseren in vivo Leukämiemodellen der Einfluß von Sulindac und NO-ASA auf
das Angehen von leukämischen Stammzellen und auf die Kinetik der
Leukämieentwicklung untersucht werden.
Dies soll neue Möglichkeiten in der Therapie der Leukämie eröffnen und
einen Beitrag dazu leiten, die bereits bekante Tumor-präventive Wirkung
von NSAID besser zu verstehen und, wenn möglich und sinnvoll, auf die
Therapie von Leukämien und andere Tumoren auszuweiten.
Veröffentlichung
July
19, 2011, DOI: 10.1371/journal.pone.0022540
nach oben zur Projektseite home
Die Bedeutung des aberrant aktivierten "stem cell Factor" (SFC) Signalwegs für die Pathogenese der akuten myeloischen Lejukämie
PD Dr. Martin Ruthardt, Med. Klinik III, Univ.-Klinikum Frankfurt am Main
Viele der Mechanismen der
Ausreifungsstörung sind bekannt, aber nichts ist darüber bekannt, wie es
zur Stimulierung der Proliferation der Stammzellen kommt und welche
Rolle sie für die Leukämo-genese spielt.
Bisher konnten wir zeigen, daß der Effekt der Translokationsprodukte von
einem Faktor abhängig zu sein scheint, der die Proliferation auch von
normalen hämopoetischen Stammzellen steuert. Dabei handelt es sich um
einen biologischen Schalter an der Zelloberfäche der Stammzellen, dem
sog. c-Kit, der durch die Bindung an ein Protein, dem sog. "stem cell
factor" (SCF), angeschaltet wird. Diese Aktivierung von c-Kit führt zur
Proliferation von normalen Stammzellen. Nun hat sich ergeben, daß
leukämische Stammzellen, viel mehr c-Kit auf ihrer Zelloberfläche haben
als normale Stammzellen.
In diesem Projekt soll geklärt werden, ob diese erhöhte Menge von c-Kit
für die Erhaltung der Leukämie von Bedeutung ist. Es gibt Medikamente,
wie das Imatinib (Gleevec), die in der Lage sind, die Aktivierung von
c-Kit zu hemmen. Sollte das aktivierte c-Kit tatsächlich von Bedeutung
für die Erhaltung der Leukämie sein, indem es die Blastenmasse immer mit
Nachschub beliefert, wäre es theoretisch möglich, zumindest diesen
Aspekt der Leukämie pharmakologisch zu behandeln.
Diese Studien stellen einen Beitrag zur Vertiefung des Verständnisses
der Mechanismen der Leukämogenese dar und sollen neue Möglichkeiten
molekularer Ansätze der Tumortherapie aufdecken, insbesondere vor dem
Hintergrund der Verfügbarkeit verschiedenster Inhibitoren, die in der
Lage sind, die Aktivierung von c-Kit zu blockieren.
siehe auch: www.spandidos-publications.com/ijo/34/6/1521
nach oben zur Projektseite home
Monitoring der Differenzierungstherapie um Valproinsäure und all-trans Retinsäure bei Patienten mit akuter myeloischer LeukämieDr. Gesine Bug, Med. Klinik III, Joh. Wolfgang Goethe-Universität Frankfurt a. M.
Die akute myeloische Leukämie (AML) ist eine aggressive Krebserkrankung, die vor allem ältere Menschen über 64 Jahre trifft. Ohne Behandlung überleben die meisten Patienten nur wenige Monate, weil die normale Blutbildung im Knochenmark zunehmend von funktionsuntüchtigen Leukämiezellen verdrängt wird. Die Patienten sterben an schweren Infektionen oder Blutungen. Wichtigstes Therapieziel ist folglich die Wiederherstellung einer ausreichenden normalen Blutbildung im Knochenmark.
Für jüngere Patienten besteht die Standardtherapie in einer intensiven Chemotherapie und evtl. einer Stammzelltransplantation, die bei älteren Patienten aufgrund der erheblichen Nebenwirkungen häufig nicht anwendbar ist. Alternative Therapiekonzepte werden deshalb dringend benötigt. In der Medizinischen Klinik III wird zur Zeit die Wirkung einer innovativen Strategie zur Leukämiebehandlung geprüft: die Differenzierungstherapie mit Valproinsäure und all-trans Retinsäure (ATRA).
Warum eine Therapie mit Valproinsäure und ATRA?
Die akute myeloische Leukämie (AML) ist eine Erkrankung der blutbildenden Stammzelle. Beim gesunden Menschen leitet sich das gesamte Blut- und Immunsystem von einer kleinen Anzahl unreifer Stammzellen im Knochenmark ab, die unter fortwährender Teilung und Ausreifung die funktionstüchtigen Blut- und Immunzellen (rote und weiße Blutkörperchen, Blutplättchen) hervorbringen. Bei der AML führen Veränderungen im Erbgut dieser Stammzellen über eine gesteigerte Teilungsrate und einen Ausreifungsstop zu einer massiven Vermehrung einer unreifen Zellpopulation, den Leukämiezellen, die die normale Blutbildung im Knochenmark verdrängen. Neue Forschungsergebnisse zeigen, daß eine Kombination von Valproinsäure und ATRA zu einer Ausreifung der Leukämiezellen in normale Blutzellen führen kann.
Ziel dieses völlig neuartigen Therapiekonzeptes ist, die Leukämie im Knochenmark zurückzudrängen und unter Kontrolle zu halten. Da Valproinsäure und ATRA auch bei längerer Einnahme sehr gut verträglich sind, eignet sich diese Tablettentherapie insbesondere für ältere Patienten, die eine intensive Chemotherapie nicht vertragen können.
nach oben
zur Projektseite
home
04/1
Finanzierung einer
Transplantationsassistenz für den Bereich Knochenmarkstransplantation
PD Dr. Hans
Martin, Med. Klinik III, Klinikum der J. W. Goethe-Universität Frankfurt
am Main
04/2
Sachmittelbeihilfe zur Erweiterung der vorhandenen GenChip Anlage zur
Analyse von Genexpressionsprofilen (Mitfinanzierung eines GeneChip
Scanner 3000) zur Verbesserung der Genexpressions-Untersuchungen
PD
Dr. Martin Ruthardt Med. Klinik III der J. W. Goethe-Universität
Frankfurt am Main
nach oben
zur Projektseite
home
04/3
Monitoring der Differenzierungstherapie mit
Valproinsäure und all-trans
Retinsäure bei Patienten mit akuter myeloischer Leukämie
Dr. Gesine Bug, Med. Klinik III, Joh. Wolfgang Goethe-Universität
Frankfurt a. M.
Die akute myeloische Leukämie (AML) ist eine aggressive Krebserkrankung,
die vor allem ältere Menschen über 64 Jahre trifft.
Ohne Behandlung überleben die meisten Patienten nur wenige Monate, weil
die normale Blutbildung im Knochenmark zunehmend von
funktionsuntüchtigen Leukämiezellen verdrängt wird. Die Patienten
sterben
an schweren Infektionen oder Blutungen.
Wichtigstes Therapieziel ist folglich die Wiederherstellung einer
ausreichenden normalen Blutbildung im Knochenmark.
Für jüngere Patienten besteht die Standardtherapie in einer intensiven
Chemotherapie und evtl. einer Stammzelltransplantation, die bei älteren
Patienten aufgrund der erheblichen Nebenwirkungen häufig nicht anwendbar
ist. Alternative Therapiekonzepte werden deshalb dringend benötigt.
In der Medizinischen Klinik III wird zur Zeit die Wirkung einer
innovativen Strategie zur Leukämiebehandlung geprüft: die
Differenzierungstherapie mit Valproinsäure und all-trans Retinsäure
(ATRA).
Warum eine
Therapie mit Valproinsäure und ATRA?
Die akute myeloische Leukämie ( AML ) ist eine Erkrankung der
blutbildenden Stammzelle. Beim gesunden Menschen leitet sich das gesamt
Blut- und Immunsystem von einer kleinen Anzahl unreifer Stammzellen im
Knochenmark ab, die unter fortwährender Teilung und Ausreifung die
funktionstüchtigen Blut- und Immun-zellen (rote und weiße
Blutkörperchen, Blutplättchen) hervorbringen.
Bei der AML führen Veränderungen im Erbgut dieser Stammzellen über eine
gesteigerte Teilungsrate und einen Ausreifungsstop zu einer massiven
Vermehrung einer unreifen Zellpopulation, den Leukämiezellen, die die
normale Blutbildung im Knochenmark verdrängen.
Neue Forschungsergebnisse zeigen, daß eine Kombination von Valproinsäure
und ATRA zu einer Ausrei-fung der Leukämiezellen in normale Blutzellen
führen kann.
Ziel dieses völlig neuartigen Therapiekonzeptes ist, die Leukämie im
Knochenmark zurückzudrängen und unter Kontrolle zu halten. Da
Valproinsäure und ATRA auch bei längerer Einnahme sehr gut verträglich
sind, eignet sich diese Tablettentherapie insbesondere für ältere
Patienten, die eine intensive Chemotherapie nicht vertragen können.
nach oben
zur Projektseite
home
04/4
W
Dr. Gesine BUG, med. Klinik III, der Johann Wolfgang Goethe-Universität, Frankfurt a. M.
Fortsetzungsförderung
Ältere Patienten mit akuten Leukämien haben trotz intensiver Chemotherapie weiterhin eine sehr geringe Chance, von der Erkrankung geheilt zu werden. Eine Verbesserung der Leukämietherapie ist durch den Einsatz neuer molekularer Medikamente zu erwarten, die spezifisch auf Leukämiezellen wirkt, ohne die normale Blutbildung zu zerstören. Eine vielversprechende Substanzgruppe stellen Histondeacetylasein-hibitoren wie die Valproinsäure (VPA) dar. In Laborversuchen bewirkt Valproinsäure eine Umwandlung der unreifen Leukämiezellen in reife Blutzellen.
In
unserer klinischen Studie war die Ansprechrate mit 10% der
Leukämiepatienten deutlich geringer als aufgrund der Laborversuche
erwartet. Dennoch profitierte eine kleine Untergruppe von Patienten von
dieser gut verträglichen und ambulant durchführbaren Therapie, wobei die
Zeit bis zum Fortschreiten der Erkrankung bis zu 8 Monate betrug. VPA
wird zudem bereits in größeren klinischen Studien in Kombination mit
Chemotherapie eingesetzt.
Ziel des Antrages ist, den Wirkmechanismus von VPA genauer zu
untersuchen und herauszufinden, für welche Leukämiepatienten diese
Therapie geeignet ist und warum sie bei anderen Patienten versagt.
Ergebnis veröffentlicht in:
http://onlinelibrary.wiley.com/doi/10.1002/cncr.21589/abstract
nach oben
zur Projektseite
home
04/5
Hochdosis-Melphalan mit autologem
Stammzellsupport bei Patienten mit
rezidivierter akuter myeloischer Leukämie (AM
PD Dr. Hans Martin und Dr. Gesine Bug, Med. Klinik III, Klinikum der J.W. Goethe Universität Ffm.
Unterstützung der klinischen Studie,
Die chemotherapeutische Behandlung von Patienten mit akuter myeloischer
Leukämie (AML) führt in vielen Fällen zur vollständigen Heilung. Kann
die Erkrankung aber durch Chemotherapie nicht beherrscht werden, kommt
es zu einem Rückfall (Rezidiv) der AML. Im Knochenmark des Patienten
können wieder Leukämie-zellen nachgewiesen werden.
In dieser Situation bietet eine allogene Stammzelltransplantation, d.h.
eine Übertragung von Blut- und Immunzellen eines passenden gesunden
Spenders, die größte Chance auf Heilung. Sie sollte zu einem Zeitpunkt
durchgeführt werden, zu dem das Knochenmark möglichst frei von
Leukämiezellen ist. Deshalb benötigen Patienten mit einem Rezidiv der
AML vor der allogenen Stammzelltransplantation zunächst eine weitere
Chemotherapie.
Eine
Standardchemotherapie steht für die rezidivierte AML bisher nicht zur
Verfügung. Aus diesem Grund wird in der Medizinischen Klinik III der
Johann Wolfgang Goethe-Universität in Zusammenarbeit mit der
Medizinischen Hochschule Hannover eine klinische Prüfung durchgeführt,
bei der 20 Patienten mit rezidivierter AML eine hochdosierte
Chemotherapie mit Melphalan erhalten. Melphalan hat eine breite
Wirksamkeit gegen verschiedene Leukämieformen und kann auch intensiv
vorbehandelten Patienten sicher verabreicht werden. Durch eine
Behandlung mit hochdosiertem Melphalan wird das Knochenmark mit den
blutbildenden Stammzellen weitgehend funktionsunfähig, so daß bis zur
Erholung der normalen Blutbildung mindestens 4 – 6 Wochen vergehen
würden. Aus diesem Grund bekommen die Patienten im Anschluß an die
Chemotherapie ihre vor einiger Zeit entnommenen und zwischenzeitlich in
flüssigem Stickstoff gelagerten eigenen (autologen) Stammzellen zurück.
Das Hauptziel dieser klinischen Prüfung besteht darin, die Sicherheit
und Verträglichkeit von Hochdosis-Melphalan und autologem
Stammzellsupport bei rezidivierten AML-Patienten unter
Berücksichtigung von sowohl kurzfristigen als auch langfristigen
Nebenwirkungen zu untersuchen. Außerdem soll die Effektivität
der Therapie mit
Hochdosis-Melphalan und autologem Stammzell-support für das
Verschwinden der Leukämiezellen und das Überleben der Patienten bestimmt
werden.
Die Erwartung eines Ansprechens der Patienten auf diese Therapie basiert vor allem auf Ergebnissen einer Pilotstudie, in der acht Patienten mit einem Rezidiv der AML mit Hochdosis-Melphalan und autologem Stammzellsupport behandelt wurden. Bei allen Patienten waren nach der Therapie keine Leukämiezellen mehr im Knochenmark nachweisbar (komplette Remission). Hochdosis-Melphalan war selbst bei den Patienten effektiv, die wenige Wochen zuvor mit einer herkömmlichen Rezidiv-chemotherapie keine komplette Remission erreicht hatten. Bei den meisten Patienten konnte anschließend eine allogene Stammzelltransplantation durchgeführt werden.
nach oben zur Projektseite home
04/6
Regulation der Differenzierung
hämatopoetischer Stammzellen durch
DNA-Methylierung von zellzyklusspezifischen Genen
Prof. Dr. Wolf-Karsten Hofmann, Med. Klinik III,
Hämatologie, Onkologie, Transfusionsmedizin,
Campus Benjamin Franklin, Charité – Universitätsmedizin Berlin
Das myelodysplastische Syndrom (MDS) ist eine Erkrankung der Blutbildung, bei der es zu einem Mangel an roten Blutkörperchen (Blutarmut – Anämie), weißen Blutkörperchen (Abwehrzellen – Granulozyten) und Blutplättchen (Thrombozyten) kommt. Diese Erkrankung beginnt allmählich und kann über viele Jahre unbemerkt verlaufen. Es kommt dann zu klinischen Symptomen (Schwäche, Abgeschlagenheit, gehäufte Infektionen, Blutungen bei Bagatellverletzungen), die den Patienten dann das erste Mal zum Arzt führen. In der Regel sind Patienten jenseits des 60. Lebensjahres von einem MDS betroffen, allerdings erkranken zunehmend junge Patienten, auch Kinder.
Eine sehr
unangenehme Eigenschaft des MDS ist, daß es die Vorstufe für eine
schwere Blutkrebserkran-kung ist, die akute myeloische Leukämie (AML).
Mehr als die Hälfte aller Patienten, die an einem MDS erkranken,
entwickeln in einem Zeitraum von 6 Monaten bis zu 10 Jahren eine solche
AML.
Am Beispiel des MDS konnte bisher durch verschiedene biologische und
genetische Methoden gezeigt werden, daß für den Verlauf der Erkrankung
von der einfachen Blutbildungsstörung bis hin zur akuten Leukämie die
Ansammlung einer Vielzahl von endogenen und exogenen Ereignissen
erforderlich ist. Schrittweise wird so die sog. Blutstammzelle
(„Blut-Mutterzelle“) geschädigt, bis die Schädigung so umfangreich ist,
daß diese Zellen bösartig entarten. Auf der Suche nach den Veränderungen
in den Stammzellen haben wir in unseren Vorarbeiten gefunden, daß es
während der Entwicklung der Blutstammzellen beim MDS zur Unterdrückung
von bestimmten Genen, die für die normale Zellteilung verantwortlich
sind (sog. Zellteilungs-Kontrollgene) kommt.
Ziel des vorliegenden Projektes ist es, diese
Störung genau zu charakterisieren und herauszufinden, zu welchem
Zeitpunkt der Ausreifung der Blutzellen diese Gene durch die Krankheit
unterdrückt werden.
Damit soll es möglich sein, dem (oder den) Defekt(en) der Blutstammzelle
beim MDS näher zu kommen. Weiterführend bietet die Analyse dieser
Krankheitsmechanismen die Möglichkeit, gezielt Medikamente gegen diese
Defekte zu entwickeln, die eine individuelle Behandlung von Patienten
mit MDS oder Leukämie ermöglichen.
siehe auch Veröffentlichung
"DNA methylation profiling of myelodysplastic syndrome hematopoietic progenitor cells during in vitro lineage-specific differentiation" in Exp Hematol. 2007 May;35(5):712-23.nach oben zur Projektseite home
04/7
Die leukämische Stammzelle als Target
molekularer Therapieasätze
PD Dr. Martin Ruthardt, Med. Klinik III der J. W.
Goethe-Universität Frankfurt a. M.
In den letzten Jahren hat die Wissenschaft überraschende Erkenntnisse über das Verhalten von Krebs-zellen, insbesondere Leukämiezellen gewonnen: Nicht alle bösartigen Zellen sind in der Lage, sich unkontrolliert zu vermehren. Im Fall der Leukämie bedeutet das, daß nur ein kleiner Teil der entarteten Leukämie-Zellen überhaupt eine Ausbreitung der Krankheit bewirken kann.
Diese sogenannten Leukämie-Stammzellen sind mit den
heute zur Verfügung stehenden konventionellen Chemotherapeutika nur sehr
schwer zu eliminieren. Weiterhin ist es weitgehend unbekannt, wie die
meisten modernen molekularen Therapien auf diese Leukämie-Stammzellen
wirken. Das ist aber von grundlegender Bedeutung für die
Erfolgsaussichten der Leukämie-Therapie, da von den wenigen
Leukämie-Stammzellen ein Rückfall der Krankheit ausgeht.
Ziel des Projekts ist es, zu klären, wie verschiedene neuartige
Substanzgruppen, die z.Zt. in die Therapie der Leukämie eingeführt
werden, auf die Leukämie-Stammzellen wirken. Es wird untersucht, wie
wirksam die heute zur Verfügung stehenden Medikamente gerade gegen die
Leukämie-Stammzellen wirken. Nur wenn es gelingt, das Wachstum dieser
Stammzellen zu verhindern, kann eine Therapie langfristig erfolgreich
sein. Denn das Ziel ist, die Leukämiepatienten dauerhaft zu heilen.
Die in dem Projekt gewonnenen Erkenntnisse sollen helfen, in Zukunft bei
der Entwicklung von Medikamenten und Therapien gegen Leukämie vermehrt
solche Substanzen einzusetzen, die besonders wirksam gegen
Leukämie-Stammzellen sind.
nach oben zur Projektseite home
04/8
Erholung der zellulären Immunfunktion gegen CMV- und EBV- Infektionen
nach Stammzelltransplantation bei Kindern und Erwachsenen
Dr. Ulrike Köhl, Klinik für Kinderheilunde III, Pädiatrische
Hämatologie, Onkologie und Hämostaseologie, Klinikum der J.W. Goethe
Universität Ffm.
Die Knochenmark-Stammzelltransplantation von Fremdspendern ist eine wirkungsvolle Therapie bei manchen Kindern und Erwachsenen mit Leukämie. Aus dem transplantierten Knochenmark entwickelt sich im Laufe von Monaten ein neues Immunsystem, dem es gelingt, die Leukämiezellen zu bekämpfen. Allerdings ist dieses sich neu entwickelnde Immunsystem erst nach und nach in der Lage, infektiöse Erreger - Bakterien, Pilze und Viren - schnell und effizient zu bekämpfen.
Im Labor für Stammzelltransplantation und
Immuntherapie der Uniklinik Frankfurt wurde im letzten Jahr dank der
Unterstützung der Gutermuth-Stiftung eine verbesserte Methode zur
Messung der Immunrekonstitution nach Stammzelltransplantation
entwickelt. Darauf aufbauend soll im Verlauf des ersten Jahres nach
Transplantation regelmäßig und engmaschig der Anteil der verschiedenen
Immunzellarten im Blut gemessen werden. An mehreren Zeitpunkten soll
zudem die Reaktivität des Abwehrsystems gegen Viren, Pilze und
Leukämiezellen getestet werden (Grafik.) Die gemessene Aktivität soll
mit der Zunahme der verschiedenen Zellen des Immunsystems verglichen
werden. Ergänzend soll untersucht werden, ob besondere genetische
Merkmale ("Polymorphismen") mit einer verminderten Immunabwehr gegen
Infektionen oder auch Leukämie-zellen zusammenhängt. Im Anschluß an die
Datenerhebung erfolgt eine statistische Auswertung, um nach Möglichkeit
„Hochrisikopatienten“ zu beschreiben. Die Ergebnisse sollen verwendet
werden, die Therapie der nachfolgenden Patienten weiter zu verbessern.
Zwischenbericht an die Gutermuth-Stiftung
vom 17.10.06
Für viele Kinder und erwachsene Patienten mit Leukämie ist die Durchführung einer Hochdosis-Chemo-therapie mit anschließender Stammzelltransplantation von einem Fremdspender oft die einzige Möglichkeit die Heilungsrate zu verbessern. Nach Transplantation regeneriert sich ein völlig neues Immunsystem aus den transplantierten Stammzellen des Spenders. Die Zeitdauer dieser Zellerholung (medizinischer Begriff: Immunrekonstitution), die volle Funktionstüchtigkeit der Immunzellen sowie das Verhältnis der verschiedenen Zellarten zueinander scheinen entscheidend die Infektionsabwehr und den Anti-Leukämie-Effekt gegen noch vorhandene Leukämiezellen zu beeinflussen.
In einem laufenden Projekt, das
ebenfalls durch die Gutermuth-Stiftung unterstützt wird, wird die
Erholung des zellulären Immunsystems nach Stammzelltransplantation in
einem engmaschigen durchfluß-zytometrischen Monitoring über z.T. mehrere
Jahre gemessen und statistisch ausgewertet1-4. In einer
ersten Auswertung, die helfen soll „Hochrisikopatienten“ mit Hilfe der
Rekonstitution einzelner Zellsubpopulationen zu definieren, war der
Marker für zytotoxische T-Zellen
signifikant für eine Trennung von „Hoch – und Niedrig-Risiko-patienten“.
Untersuchungen über die Erholung des Immunsystems in der Abwehr gegen Viren sind technisch schwierig und ergaben lange Zeit unbefriedigende Ergebnisse. Es ist aber im Stammzell-Labor vor kurzem gelungen auf der Basis von Tetramerbestimmungen einen Assay zu entwickeln, der die genaue Bestimmung von CMV- und EBV-spezifischen T-Zellen erlaubt. Besonders die Cytomegalievirus-Infektion stellt ein großes Problem nach Transplantation dar.
Im vorliegenden Antrag konnten bisher bei 17 pädiatrischen und bei 17 internistischen Patienten die Entwicklung dieser CMV-spezifischen T-Zellen untersucht werden. Dabei zeigte sich, dass Patienten, die CMV-spezifische T-Zellen entwickeln in den meisten Fällen die Cytomegalievirus-Infektion nach Transplantation überwinden können. Der Anteil dieser CMV-spezfischen T-Zellen steigt bei diesen Patienten nach Stammzelltransplantation kontinuierlich an (Beispiel siehe Abb. 1A+B). Patienten, die keine solchen CMV-spezifische T-Zellen entwickeln, leiden zum überwiegenden Teil an schwer verlaufenden Cytomegalievirus-Infektionen.
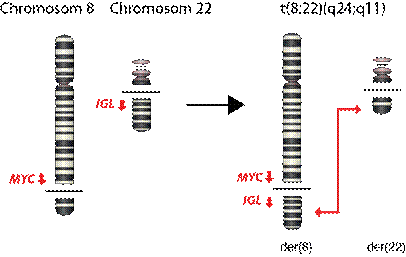
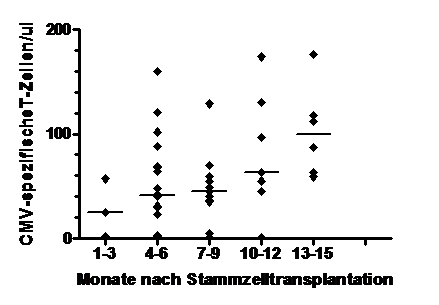
Abb. 1: Entwicklung CMV-spezifischer T-Zellen
nach Stammzelltransplantation.
Im erste Bild ist der Verlauf eines Patienten nach Stammzelltransplantation (SZT) gezeigt. Am Tag 11 nach SZT entwickelt dieser Patient eine CMV-Infektion, die neben dem klinischen Bild an Hand seiner Virus-Last (CMV-DNA-Kopienzahl, rechte Y-Achse; Kurve mit offenen Quadraten) bestimmt wurde. Ab Tag 26 nach SZT konnte bei diesem Patienten zum ersten Mal CMV-spezifische T-Zellen gegen CMV nachgewiesen werden, die dann täglich anstiegen (linke Y-Achse, Kurve mit dicken ausgefüllten Kreisen). Mit Zunahme seiner CMV-spezifischen T-Zellen verschwand die CMV-Infektion, sichtbar auch am Rückgang der Virus-Last. Die dünnen Kurven zeigen die zelluläre Erholung seiner verschiedenen T-Zellen.
Im zweiten Bild ist von allen 22 der 34 untersuchten Patienten, die CMV-spezfische T-Zellen bildeten, die Entwicklung dieser antigenspezifischen Zellen dargestellt.
siehe Veröffentllichung:
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3521740/pdf/pone.0050248.pdf
Eigene Literatur
1. Koehl U, Sörensen J, Esser R, Zimmermann S, Grüttner HP, Tonn T, Seidl C, Seifried E, Klingebiel T, Schwabe D. Blood Cells, Mol. and Diseases 33: 261-266 (2004)
2.
Beck O,
Seidl C, Lehrnbecher T, Kreyenberg H,
Schwabe D, Klingebiel T
Seifried E, Bader P,
Koehl U.
Eur J Hematol 76:237-44 (2006).
3. Ott MG, Schmidt M, Schwarzwaelder K, Stein S, Siler U, Koehl U, Glimm H, Kühlcke K, Schilz A, Kunkel H, Naundorf S, Brinkmann A, Deichmann A, Fischer M, Ball C, Pilz I, Trasher A, Hoelzer D, Kalle C, Seger R, Grez M. Nature Medicine 12(4):401-9 (2006) Passweg JR, Koehl U, Uharek L, Meyer-Monrad M, Tichelli A. Best Pract + Res Clin Haematol 19: 811-24 (20
siehe Veröffentlichungen:
Multimer monitoring of CMV-specific T cells in research and in clinical applications;
Scand J. Immunol. 2013 Mar;77/(3):213-20. doi:10.1111/sji. 12024;
Reconstitution of cytomegaloviris specific T cells afer pediatric allogeneic stem cell transplantation:
reults from a pilot study using a multi-allele CMV tetramer group Klin Padiatr.
2008 Nov-Dec;220(6):348-52. doi: 10.1055/s-0028-1086029. Epub 2008 Oct 23.
nach oben
zur Projektseite
home
04/9
Therapie der Steroid
refraktären akuten Graft versus Host Disease
Dr. Stefan A. Klein, Klinikum Bayreuth, Preuschwitzer Str. 101,
(früher med. Klinik III der
J. W. Goethe-Universität Frankfurt a. M.)
Die Alfred und Angelika Gutermuth-Stiftung unterstützt mit diesem Projekt die Durchführung einer multizentrischen, prospektiven und randomisierten Studie zur Therapie der Steroid refraktären Graft versus Host Disease (GvHD) nach allogener hämatopoetischer Zelltransplantation.
Die akute GvHD ist die Haupttodesursache nach allogener Knochenmarktransplantation. Es handelt sich dabei um eine von weißen Blutkörperchen des Spenders, den sogenannten T-Lymphozyten, vermittelte Reaktion gegen Gewebe des Empfängers. Besonders betroffen von dieser Reaktion ist der Darm. Die Patienten leiden unter schwersten Durchfällen und Bauchschmerzen. Bei schwerwiegenden Verläufen kann es zum Absterben von Darmgewebe oder zu tödlichen vom Darm ausgehenden Infektionen kommen. Die Gabe von hochdosiertem Kortison ist als Standardtherapie der akuten GvHD etabliert. Leider sprechen insbesondere bei schwerwiegenden Verläufen nur weniger als die Hälfte der Patienten auf die Kortison-therapie an. Bei Versagen der Kortisontherapie ist die Wahrscheinlichkeit, daß der Patient an der GvHD verstirbt, sehr hoch. Für Patienten ohne Ansprechen auf Kortison, d.h. bei Vorliegen einer sogenannten Steroid refraktären GvHD, gibt es bislang keine etablierte Therapie. Weltweit existiert kein Therapieprotokoll, für das gegenüber dem alleinigen Einsatz von Kortison, eine Verbesserung der Überlebensrate belegt ist. Das Fehlen eines Standardvorgehens hat dazu geführt, daß jedes Transplantationsteam ein hauseigenes Protokoll ohne jeden wissenschaftlichen Beleg der Wirksamkeit einsetzt.
Als
Grundlage für dieses Projekt diente eine von der Gutermuth-Stiftung
unterstützte Pilotstudie (2001), in der wir die Wirksamkeit von
Pentostatin als Medikament zur Therapie der akuten Graft versus Host
Disease (GvHD) belegen konnten. Bei fast allen Patienten wurden die
Symptome der GvHD durch Pentostatin deutlich vermindert. Bei etwa ¾ der
Patienten verschwanden die GvHD-Symptome gänzlich. Auch die
Überlebensrate der Patienten war überdurchschnittlich gut. Diese
ermutigenden Ergebnisse veranlassten uns, eine randomisierte Studie zur
Prüfung der Effektivität der Pentostatintherapie aufzulegen.
Das Ziel der jetzt laufenden prospektiven, multizentrischen und
randomisierten Studie ist es zu belegen, dass eine standardisierte
Therapie mit Pentostatin die Überlebensrate von Patienten mit akuter
Steroid refraktärer GvHD verbessert.
In die Studie eingeschlossen werden Patienten, bei denen es nach dem
Einsatz von hochdosiertem Kortison nicht zu einer Besserung gekommen
ist. In der Studie erfolgt eine Randomisierung in zwei unterschiedliche
Therapiearme. Im Kontrollarm werden Patienten lediglich mit Kortison, im
Prüfarm zusätzlich mit Pentostatin behandelt. Insgesamt sollen 100
Patienten (50 pro Therapiearm) in die Studie eingeschlossen werden.
Die Studie ist derzeit an den Unikliniken
Dresden, Heidelberg, Frankfurt und Münster, sowie an der Deutschen
Klinik für Diagnostik in Wiesbaden aktiv. Weitere Zentren haben auf der
Studiensitzung der kooperativen Transplantationsstudiengruppe im Februar
ihre Teilnahmeabsicht bekundet. Bislang sind 15 Patienten in die Studie
eingeschlossen. Da es sich um eine prospektive und randomisierte Studie
handelt, können die bisherigen Therapieergebnisse in den beiden Armen
noch nicht veröffentlicht werden.
Bundesweit findet einzig diese Studie bei akuter GvHD Akzeptanz in
mehreren Transplantationszentren.
Die Durchführung der Studie wird entscheidend mit Hilfe der
Unterstützung durch die Alfred und Angelika Gutermuth-Stiftung
ermöglicht. Die Unterstützung hilft, Diagnostik und Dokumentation in den
teilnehmenden Zentren zu verbessern. Wir haben die Hoffnung mit dieser
Studie die Therapie der Steroid refraktären akuten GvHD wesentlich
voranzubringen und so die Prognose nach allogener
Stammzelltransplantation ein gutes Stück zu verbessern.
siehe
auch:
2006/1 Bedeutung
der abdominellen Sonographie in der Diagnostik der Graft versus Host Disease
nach oben
zur Projektseite
home
geförderte Projekte 2005
05/1
Untersuchung der Expression der dNa (Cytosin-5) – Methyltransferasen 1,
3a und 3b (DNMT1, 3A und 3B) während der in-vitro Differenzierung
von hämatopoe-tischen Stammzellen von Patienten mit myelodysplastischem
Syndrom
Prof. Dr. Wolf-Karsten Hofmann, Med. Klinik III, Hämatologie, Onkologie, Transfusionsmedizin, Campus Benjamin Franklin, Charité – Universitätsmedizin Berlin
In
den letzten Jahren ist es gelungen, verschiedene Defekte in den
Blutzellen bei Patienten mit myelodys-plastischem Syndrom (MDS)
nachzuweisen. Dabei hat man erkannt, daß das gestörte Wachstum dieser
Zellen ursächlich mit Veränderungen im Bereich der Chromosomen oder aber
einzelner Gene verknüpft ist. Dies ist auch die Ursache dafür, daß es
später zur Entwicklung der bösartigen akuten myeloischen Leukämie kommt.
Eine große Anzahl von Störungen liegt im Bereich der Gen-Informationen,
die in der menschlichen DNA gespeichert sind, vor. Es gibt verschiedene
Veränderungen wie den Verlust von DNA-Abschnitten (Deletionen),
Veränderungen des genetischen „Codes“ in den MDS-Zellen (Mutationen),
Vervielfachungen von DNA-Abschnitten (zum Beispiel Verdopplungen –
Duplikationen) und andere. Außerdem hat man durch systematische
Untersuchungen herausgefunden, daß einige Abschnitte der DNA auf eine
bestimmte Art und Weise chemisch modifiziert („methyliert“) sind. Im
Zusammenhang mit einem Projekt, welches bereits von der
Gutermuth-Stiftung gefördert wurde, konnten solche chemisch veränderten
Abschnitte auf der DNA von MDS-Zellen gefunden werden.
Das jetzt hier beantragte Projekt setzt diese Arbeiten fort und soll die
Ursachen für diese krankhafte chemische Veränderung untersuchen. Es ist
bekannt, daß einige Körper-Enzyme (Katalysatoren) für diese
„Methylierung“ verantwortlich sind (sogenannte DNA-Methyltransferasen),
offensichtlich sind diese Enzyme beim MDS falsch gesteuert und deshalb
im Übermaß in den MDS-Zellen vorhanden. Die Folge könnte sein, daß die
DNA und die darin verschlüsselten genetischen Informationen falsch oder
gar nicht abgelesen werden, was zu einer dramatischen Störung der
Zellteilung und des Wachstums der Blutzellen führen kann. Die Folge ist
die beim MDS beobachtete Blutarmut.
Im vorliegenden Projekt soll die Konzentration verschiedener
DNA-Methyltransferasen in Blutzellen ver-schiedener Reifungsstadien beim
MDS untersucht werden. Die Ergebnisse sollen Aufschluß über mögliche
Ursachen der chemischen Veränderungen von genomischer DNA beim MDS
geben. Da sich bereits Medikamente in der Entwicklung befinden, die die
Methylierung von DNA-Abschnitten hemmen/verhindern können, sind die
Informationen aus den hier geplanten Experimenten von grundlegender
Bedeutung für die zukünftige erfolgreiche Behandlung beim MDS.
Veröffentllichung in BioTechniques, Vol. 50, No. 3, March 2011, pp. 161–164
nach oben zur Projektseite home
05/2
Identifizierung und Monitoring von minimaler
Resterkrankung bei Kindern mit akuter lymphatischer Leukämie mittels
multiparametrischer Immunphäno-typisierung
Dr. Richard Ratei und Dr. Leonid Karawajew aus der Forschungsgruppe von Prof. Wolf-Dieter Ludwig (Robert-Rössle-Klinik, Charité – Universitätsmedizin Berlin/Helios Klinikum Berlin und Max-Delbrück-Centrum für Molekulare Medizin, MDC, Berlin-Buch)
Bei der
Behandlung akuter lymphatischer Leukämien ist die rasche und
vollständige Eliminierung des leukämischen Zellklons von großer
Bedeutung für die Heilung. Unter Therapie können resistente leukämische
Zellen im Knochenmark persistieren (= minimale Resterkrankung,
MRD) und selbst in sehr geringer Anzahl zu einem Rückfall der Erkrankung
führen. Mit der frühzeitigen Erkennung einer persistierenden
Zellpopulation kann die Therapie rechtzeitig umgestellt und intensiviert
werden, um trotzdem eine Heilung für den Patienten zu erreichen.
Für die Beurteilung des Behandlungserfolges wird traditionell die
mikroskopische Begutachtung des Knochenmarkes, allerdings mit dem
Nachteil der geringen Nachweisempfindlichkeit von nur einer
Leukämiezelle in 100 normalen Zellen, herangezogen. Moderne
molekularbiologische und durchfluß-zytometrische Methoden zeigen hier
eine wesentliche höhere Empfindlichkeit und Genauigkeit und haben sich
deshalb zum MRD-Nachweis bei der Behandlung der akuten lymphatischen
Leukämien als ein entscheidender prognostischer Parameter für das
Rezidivrisiko erwiesen.
Die multiparametrische Durchflußzytometrie (MFC) ist im Vergleich zu den
anderen Methoden wesentlich schneller und billiger, und bietet die
Möglichkeit der exakten MRD-Quantifizierung. Die ersten Ergebnisse haben
gezeigt, dass die etablierte Nachweismethode von MRD mittels MFC mit
einer Sensitivität von 10-3 - 10-4 bei mehr als
90% der Patienten durchführbar ist. Im Rahmen des von der Wilhelm Sander
Stiftung für zwei Jahre mit 200.000,- € geförderten Projektes wird der
prospektive, durchflußzytometrische Nachweis
von MRD bei Kindern, die im ALL –BFM 2000 Protokoll behandelt werden,
durchgeführt und hinsichtlich der prognostischen Relevanz evaluiert.
Weiterhin werden die MRD-Zellen mittels Zellsortierung und
Genexpressionsanalyse umfassend molekular-biologisch analysiert, um
somit neue diagnostische Marker und therapiespezifische Kandidatengene
identifizieren zu können.
Es wird die Finanzierung einer MTA unterstützt, zu deren
Aufgaben es u. a. gehört, im Rahmen des Forschungsvorhabens den Aufbau,
die Erweiterung und Verwaltung einer relationalen Datenbank
durchzuführen, mit der die experimentellen und klinischen Datensätze
verknüpft werden.
nach oben zur Projektseite home
05/3
Charakterisierung der
unterschiedlichen Zielzellen verschiedener
Histon-Deacetylase-Inhibitoren im Stammzellkompartiment der normalen
und leu-kämischen Hämopoese
PD
Dr. Martin Ruthardt,
M.D., Med. Klinik II/Hämatologie,Klinikum der J.W.Goethe-Univ. Ffm.
u. Dr. Ulrike Köhl, Pädiatrische Hämatologie und
Onkologie,Klinikum der J.W.Goethe-Univ. Ffm.
In
Kooperation zwischen der Hämatologie für Kinder und für erwachsene
Patienten soll der Einfluß verschiedener Histon-Deacetylase-Inhibitoren
auf Stammzellen und auf leukämische Zellen untersucht werden.
Die Histon-Deacetylase-Inhibitoren wirken auf die Ausreifung und
Vermehrung sowie auf das Absterben von normalen Stammzellen und
leukämischen Zellen sehr unterschiedlich. Daher sind diese Substanzen
sehr interessant für zukünftige Therapien.
Im Rahmen dieses Antrags sollen die Wirkmechanismen der
Histon-Deacetylase-Inhibitoren durch Genexpressions-Analysen und
Zytokin-Arrays genauer untersucht werden.
siehe auch: Cancer
Research 65, 6080-6088, July 15,
2005
nach oben zur Projektseite home
05/4
Einfluss spezifischer
Proteine des Humanen Zytomegalievirus (HCMV) auf die Progression
maligner Erkrankungen
Dr. phil. nat. Jens-Uwe Vogel, Frankfurter
Stiftung für krebskranke Kinder – Dr.-Petra-Joh-Haus -
Veröffentlilchungen weisen auf Zusammenhänge
zwischen dem humanen Zytomegalievirus (HCMV) und der Verschlechterung
von Tumorerkrankungen hin, während die Bedeutung von HCMV-Infektionen
auf die Progerssion von Leukämien bislang nur ungenügend untersucht ist.
In aktuellen Studien können bei einigen Tumoren HCMV-DNA und Proteine
mit hoher Frequenz im Tumor-gewebe nachgeiwesen werden, während das
umliegede Gewebe virusfrei ist.
Im Rahmen des Forschungsprojektes soll der generelle Einfluß
HCMN-spezifischer Proteine bei Leukämien untersucht werden.
Für die Modulation der zellulären Gene können sowohl Hüllproteine des
Virus, als auch virale Proteine, die früh im Vermehrunszyklus gebildet
werden, eine Rolle spielen. Während Hüllproteine eine zentrale Rolle bei
der Infektionausbreitung haben, wirken die frühen viralen Proteine
regulierend auf die Virusvermehrungung.
Am Modellsystem der akuten lymphatischen Leukämie (ALL) bei Kindern, der
häufigsten Krebserkran-kung im Kindesalter, soll durch
Mikroarray-Analysen der Einfluß einzelner viraler Gene auf zelluläre
Gene untersucht werden.
In Zusammenarbeit mit dem Senckenbergschen Institut für Pathologie und
der pädiatrischen Hämatologie und Onkologie des Klinikums der J. W.
Goethe-Univeersität soll dabei untersucht werden, inwiefern diese
HCMC-kodierten Proteine zu einer differentiellen Regulierung zellulärer
Gene beitragen und zu einer Modulierung der Zellprofileration,
Motilität, Adhäsion sowie Angiogenese führen.
nach oben zur Projektseite home
05/5
Effekt von Valproinsäure und
5-Azacytidin in Kombination mit intensiver Chemotherapie auf das normale
hämatopoetische Stammzellkompartiment
Dr. Gesine Bug, Med. Klinik III, Klinikum der
J.W. Goethe Universität Frankfurt a. M.
Die Standardchemotherapie und ihre Ergebnisse haben sich bei Patienten mit akuter myeloischer Leukämie (AML) in den letzten 20 - 30 Jahren kaum verbessert, weil keine neuen, wirkungsvollen Substanzen zur Verfügung standen. Gegenwärtig werden innovative molekulare Medikamente, die als Monotherapien bei einzelnen AML-Patienten eine vielver-sprechende Wirkung gezeigt haben, zunehmend mit herkömmlicher Chemotherapie kombiniert, um ihre Effektivität zu erhöhen.
Valproinsäure (VPA) und
5-Azacytidin (5'-Aza) gehören zu einer Klasse von epigenetisch wirksamen
Medikamenten, die die Ausreifung und den Zelltod funktionsloser
Leukämiezellen herbeiführen und damit eine Kontrolle der AML erreichen
können.
Wir haben in Vorarbeiten gezeigt, daß VPA und/oder 5'Aza auf gesunde
blutbildende Stammzellen im Knochenmark genau die gegenteilige Wirkung
haben: sie fördern die Erhaltung und Vermehrung der unreifen
Stammzellen.
Standardchemotherapien bewirken
immer auch eine Zerstörung der normalen Blutzellen, die aber nach Ende
der Chemotherapie aus den Stammzellen im Knochenmark durch Zellteilung
und Ausreifung in funktions-tüchtige Blutzellen nachgebildet werden.
Setzt man nun VPA und/oder 5'Aza gleichzeitig mit einer
Standardchemotherapie ein, so ist zu befürchten, daß es zu einer
Erschöpfung der normalen Blutbildung kommt, weil gesunde Stammzellen
während der Chemotherapie in die Zellteilung getrieben werden.
Da Chemotherapie u. a. in der Teilung befindliche leukämische, aber auch
normale Zellen vernichtet, könnten deutlich mehr gesund Stammzellen als
unter alleiniger Chemotherapie üblich getroffen werden und für die
anschließende Erholungder Blutbildung nicht mehr zur Verfügung stehen.
In Mausversuchen soll deshalb die Wirkung von VPA und/oder 5'Aza in Kombination mit Chemotherapie auf die normale Blutbildung genau analysiert werden.
nach oben zur Projektseite home
05/6
Mutationsanalyse von FBW7/hCDC4 in
Myelodysplastischem Syndrom (MDS) und Akuter Myeloischer Leukämie (AML)
Prof. Dr. Wolf-Karsten
Hofmann, Med. Klinik III, Hämatologie, Onkologie, Transfusionsmedizin,
Campus Benjamin Franklin, Charité – Universitätsmedizin Berlin
Beim myelodysplastischen Syndrom (MDS) und bei
der akuten myeloischen Leukämie (AML) liegt eine große Anzahl von
Störungen im Bereich der Gen-Informationen, die in der menschlichen DNA
gespeichert sind, vor.
Es gibt verschiedene Veränderungen wie den Verlust von DNA-Abschnitten
(Deletionen), Veränderungen des genetischen „Codes“ in den MDS-Zellen
(Mutationen), Vervielfachungen von DNA-Abschnitten (zum Beispiel
Verdopplungen – Duplikationen) und andere. Vor allem Veränderungen im
Bereich sogenannter „Tumor-Suppressor-Gene“ (also von Genen, welche die
Entstehung von Tumoren verhindern sollen), können für die Entstehung
eines MDS/einer AML verantwortlich sein.
Das kürzlich entdeckte Tumor-Suppressor-Gen FBW7 hat eine große Bedeutung bei der Regulation von Zellteilungs-Prozessen. Eine Störung dieser Prozesse kann zur Entstehung von Tumoren führen. Deshalb ist in normalen Zellen eine sichere Funktion dieses Gens erforderlich. Durch Untersuchung dieses Gens in soliden Tumorzellen konnte gezeigt werden, daß FBW7 in Tumorzellen verändert (mutiert) ist. Damit könnte es eine Ursache für die Tumorentstehung sein.
Im vorliegenden soll das Gen FBW7
in Knochenmarkzellen von Patienten mit MDS und AML untersucht werden.
Dadurch soll es möglich sein, Störungen dieses Gens beim MDS bzw. bei
der AML zu entdecken und damit weitere Hinweise auf die
Krankheitsentstehungen dieser Blutkrebsformen zu bekommen.
Die in diesem Projekt geplanten Arbeiten stellen weltweit die erste
Mutationsanalyse von FBW7 in Leukämiezellen dar.
Veröffentlichung in International Journal of Medical Sciences ISSN 1449-1907
Mutation Analysis of hCDC4 in AML Cells Identifies a New Intronic Polymorphism
nach oben
zur Projektseite
home
geförderte Projekte 2006
06/1
Untersuchungen zur Bedeutung der abdominellen Sonographie in der
Diagnostik der Graft versus Host Disease und wichtigen
Differentialdiagnose
PD Dr. med. Stefan A. Klein, Klinikum Bayreuth
GmbH, Medizinische Klinik IV,
Preuschwitzer Str. 101, 95445 Bayreuth, Tel: 0921 400 6302, Fax: 0921
400 6309
Fachgebiet und Arbeitsrichtung: Innere Medizin, Hämatologie,
Stammzelltransplantation, Sonographie
Gastrointestinale Komplikationen
haben einen großen Anteil an der transplantationsassoziierten Mortalität
nach einer allogenen hämatopoetischen Zelltransplantation. Die
Hauptkomplikation stellt hierbei die akute Graft versus Host Disease
(aGvHD) mit gastrointestinaler Manifestation dar. Wichtige
Differentialdiagnosen sind virale Infektionen (CMV, HSV, VZV, HHV-6 oder
Adenoviren), bakterielle Infektionen (insbesondere Clostridium
difficile) oder Schäden, die durch die Konditionierung hervorgerufen
werden. Die Therapie der aGvHD besteht in einer Verschärfung der
Immunsuppression; die der Infektionen aus einer
antibiotischen/antiviralen Therapie in Kombination mit dem Versuch einer
Reduktion der Intensität der Immunsuppression. Die therapeutischen
Ansätze sind somit geradezu gegenläufig. Für einen therapeu-tischen
Erfolg und für das Überleben des Patienten ist eine frühzeitige
zuverlässige Diagnose entscheidend. Die klinische Symptomatik alleine
besteht im Falle der meisten Differentialdiagnosen aus
Übelkeit/Erbrechen und vor allem aus Durchfällen. Im Falle des häufigen
und unspezifischen Symptoms „Durchfall“ muß rasch eine Diagnose gestellt
werden. Die diagnostische Methode der Wahl ist die endoskopische
Untersuchung des Dickdarms bis hin zum Ende des Dünndarms. Es werden
Biopsien entnommen und diese histologisch und virologisch (PCR auf
Virus-Nukleinsäuren) untersucht. Diese Verfahren sind zeitaufwendig,
sowie für den Patienten belastend und risiko behaftet. Eine weit weniger
belastende und zugleich schnell und jederzeit verfügbare Methode stellt
die abdominelle Sonographie dar.
In einer Pilotstudie konnten wir wichtige sonographische Befunde, die
typisch für eine akute Darm-GvHD sind, identifizieren. Diese
sonographischen Befunde erlauben eine diagnostische Abgrenzung gegenüber
den wesentlichen Differentialdiagnosen.
Die Ergebnisse der Pilotstudie auf der Basis von 12 untersuchten
Patienten mit akuter Darm-GvHD konnten wir 2001 im British Journal of
Haematology
115(4):929-934,publizieren (Klein et al., 2001). Als typische
sonographische Befunde bei der akuten Darm-GvHD identifizierten wir:
·
Symmetrische Verdickung der Darmwand
(>2 mm); jeder Darmabschnitt kann befallen sein,
jedoch ist am häufigsten die Ileozökalregion
betroffen.
· Bei schweren Verläufen kann es zu einer Separierung der inneren Darmwandschichten insbesondere im Bereich des Dünndarms kommen.
· Patienten mit großen Stuhlvolumina (> 1500 ml) weisen häufig eine Beteiligung des Dünndarms auf. Neben einer verdickten Dünndarmwand zeigt sich bei diesen Patienten ein der einheimischen Sprue ähnliches sonographisches Bild der sekretorischen Diarrhoe. Neben einer Dilatation der betroffenen Jejunal- oder Ileumschlingen mit ungleichmäßigen, nur noch angedeuteten und ”aufgelockert” wirkenden Kerckring’schen Falten zeigt sich eine auffällige Flüssigkeitsvermehrung im Lumen mit lebhafter Peristaltik.
·
Die duplexsonographische Untersuchung des Blutflusses in der Art.
mesenterica superior und der Perfusion der Darmwand führte zu
charakteristischen Befunden: Patienten mit normalem oder gesteigerten
Blutfluß in der Art. mesenterica superior und einer normalen oder
gesteigerten Perfusion der Darmwand sprachen auf die GvHD-Therapie an.
Patienten, bei denen eine verminderte systolische Geschwindigkeit
in der
Arteria mesenterica superior und ein Verlust des diastolischen
Flussanteils im Zusammenhang mit einer verdickten Darmwand ohne
Darstellung einer muralen Perfusion festzustellen war, sprachen auf die
immunsuppressive Therapie nicht an und verstarben.
In den vergangenen Jahren konnten wir nun die abdominelle Sonographie
bei nahezu 100 Patienten mit gastrointestinalen Symptomen anwenden.
Die
mit dieser Projektbeschreibung beantragten Mittel möchten wir zur
Auswertung der sono-graphischen Befunde, sowie zur Dokumentation der
Verläufe der untersuchten Patienten nutzen.
Die Fragestellungen des Projektes sind:
1. Sind die in der Pilotstudie erhobenen Befunde reproduzierbar und an Hand der übrigen Untersuchungs-befunde der Patienten auch validierbar?
2. Können weitere typische sonographische Befunde bei Patienten mit gastrointestinalen Symptomen nach allogener Stammzelltransplantation identifiziert und der histologisch verifizierten Diagnose zugeordnet werden?
3. Können typische sonographische Befunde im Falle von viralen Infektionen erhoben werden?
4. Gibt es zusätzlich zu den bereits beschriebenen zusätzliche Risikofaktoren, die einen für das Ansprechen auf eine Therapie prädiktiven Wert haben?
nach oben
zur Projektseite
home
06/2
Bildung eines Fonds für
Zuwendungen an Doktoranden-stipendiaten, soweit sich ihre Arbeiten im
Rahmen der satzungsgemäßen Ziele der Stiftung bewegen
Fördersumme 4.000,--€
(in Teilbeträgen zu je 2.000,-- bzw. 1.000,--
€)
a)
Strahlenbiologische Wirkung des Betaemitters Yttrium-90 auf die
Zelllinie
BV-173 im Vergleich zur 6
MeV-Photonenbestrahlung
Julia
Schwarz (Doktorand),
Med. Klinik III, Univ.-Klinikum Frankfurt a.
M.
b)
Strahlenbiologische Wirkung des Betaemitters Rhenium-86 auf die
Zelllinie
BV-173 im Vergleich zur 6 MeV-Photonenbestrahlung
Jennifer
Gessler (Doktorand), Med.
Klinik III, Univ.-Klinikum Frankfurt a. M
c)
Gastrointestinale Viruseffekte und Graft-versus-Host-Disease bei
erwachsenen Patienten nach allogener
Knochenmark- /Stammzelltrans-
plantation
Sina Hess
(Doktorand), Med.
Klinik III, Univ.-Klinikum Frankfurt a. M
Das
Ergebnis wurde veröffentlicht in PubMed Abstract
http://www.ncbi.nlm.nih.gov/pubmed/22048789
Der
Original-Artikel ist veröffentlicht in:
Ann Hematol. 2012 May;91(5):737-42. doi:
10.1007/s00277-011-1354-5. Epub 2011 Nov 3.
d)
Retrospektive Analyse
von pulmonalen Spätfolgen nach Ganzkörperbe-
strahlung bei Patienten mit hämatopoetischer Stammzellbestrahlung
Kristina
Würth (Doktorand),
Med. Klinik III, Univ.-Klinikum Frankfurt a.
M
nach oben zur Projektseite home
06/3
Charakterisierung von molekulargenetischen Veränderungen in
Stromazellen von Patienten mit myelodysplastischem Syndrom.
Prof.
Dr. Wolf-Karsten Hofmann, Med.
Klinik III, Hämatologie, Onkologie, Transfusionsmedizin,
Campus Benjamin Franklin, Charité – Universitätsmedizin Berlin
Die Ursache für die Entstehung des myelodysplastischen
Syndroms (MDS) ist bis heute nicht aufgeklärt. Offensichtlich handelt es
sich nicht – wie bei anderen Erkrankungen – um einen Einzeldefekt auf
molekularer oder zellulärer Ebene, sondern es liegt eine Kombination von
verschiedenen Störungen vor.
Bisher wurden beim MDS vor allem die Blutbildungszellen (Blut-Stammzellen) untersucht. Aus anderen hämatologischen Neoplasien (maligne Lymphome, akute Leukämien) ist jedoch bekannt, daß offensichtlich auch molekulare Defekte in Knochenmark-Bindegewebezellen vorliegen, diese Zellen möglicherweise sogar bösartig entartet sind. Beim MDS gibt es dazu bisher keine publizierten systematischen Daten zu dieser Fragestellung.
Das vorliegende Projekt soll untersuchen, ob und wie die Knochenmark-Bindegewebszellen (auch – Stromazellen genannt) von Patienten mit MDS mit in die Störung der Blutbildung einbezogen sind und entsprechende molekulare Defekte aufweisen. Wesentlicher Bestandteil des Projektes ist der Vergleich der Daten aus Stromazellen mit den Daten aus Blutstammzellen von MDS-Patienten. Dabei werden moderne Techniken der Molekulargenetik (Mikroarray-Analysen, Genom-Analysen mit sogenannten hochauflösenden DNA-Chips) angewandt.
nach oben zur Projektseite home
Aktivität von natürlichen Killer (NK-) Zellen gegen Leukämiezellen nach Stammzelltransplantation bei Kindern: Interaktion von NK-Zellen und Dendritischen Zellen (DZ) zur Steigerung der Zytotoxizität
Frau Dr. rer. nat. Ulrike Köhl, Med. Klinik II, Pädiatrische Hämatologie und Onkologie,
Klinikum der J.W.Goethe Universität Frankfurt
Für Kinder mit akuten Hochrisikoleukämien stellt die Immuntherapie mit Spenderzellen nach Stammzelltransplantation (SZT) eine wichtige Therapiemöglichkeit dar. Dabei bietet die Be-handlung mit aufgereinigten Natürlichen Killer (NK)-Zellen des Spenders die Möglichkeit einen immunologischen Effekt gegen noch verbliebene Leukämiezellen zu erzielen. Im Gegensatz zu Zelltherapien mit T-Zellen (eine andere Art von Immunzellen) sind die Nebenwirkungen durch NK-Zellen sehr gering, da das gesunde Gewebe des Patienten durch NK-Zellen so gut wie nicht angegriffen wird.
In einer ersten klinischen Studie konnten wir zeigen, dass es möglich ist, hochaufgereinigte NK-Zellen von den Eltern als Spender zu gewinnen und anschließend als Therapie bei den krebskranken Kindern einzusetzen (Koehl Mol Dis 2004 + Klin Päd 2005).
Für eine effektive Therapie mit NK-Zellen ist es notwendig, die Aktivität von NK-Zellen gegen Leukämiezellen zu steigern. Im vorliegenden Antrag sollen daher die zellulären Wirkmechanismen, die zu einer verstärkten NK-Zell-Zytotoxizität gegen Leukämiezellen führen, immunologisch und molekularbiologisch genau charakterisiert werden.
Dazu wird der Einfluss verschiedener Zytokine (Botenstoffe, die NK-Zellen stimulieren können) und die Interaktion zu den so genannten Dendritischen Zellen, die eine Schlüsselrolle in der Aktivierung von NK-Zellen einnehmen, untersucht. Die Hochregulierung unterschiedlicher Rezeptoren auf der Oberfläche von NK-Zellen, die mit einer Zunahme der zytotoxischen Aktivität gegenüber Leukämiezellen zusammenhängt, wird bestimmt. Die Untersuchungen erfolgen zunächst an Zelllinien, später an malignen Leukämiezellen unserer Patienten.
Die Ergebnisse sollen beitragen, die
Immuntherapie mit NK-Zellen nach Stammzelltransplantation zu optimieren
und damit die Heilungschance der Kinder mit Hochrisikoleukämien zu
verbessern.
nach oben
zur Projektseite
home
06/5
Die parakrine Peptidtransduktion als
therapeutischer Ansatz für die
Philadelphia Chromosom-positive Leukämie
PD Dr. Martin
Ruthardt, Med. Klinik II / Hämatologie Klinikum der J.W. Goethe
Universität Frankfurt
Die Leukämie hat ihre
Ursache in der bösartigen Veränderung einzelner Zellen im Knochenmark.
Je nachdem ob die Leukämiezellen aussehen wie Lymphozyten oder wie
Nicht-Lymphozyten spricht man jeweils von lymphatiser oder myeloischer
Leukämie.
Je nach Krankheitsverlauf werden diese beiden Gruppen noch in chronisch
und akut unterteilt. Die akuten Leukämien führen unbehandelt innerhalb
von Tagen oder weniger Wochen zum Tod, während die chronischen Leukämien
einen längeren Verlauf aufweisen.
Die Unterscheidung sowohl zwischen chronisch und akut, als auch zwischen
lymphatisch und myeloisch ist von großer Bedeutung, da dies für die zu
verabreichende Therapie und die Prognose der Patienten entscheidend ist.
Bei über 60 % der Leukämien werden genetische Aberrationen für die
Leukämogenese verantwortlich gemacht. Die häufigsten genetischen
Aberrationen sind Chromosomentrnaslokationen, bei denen Stücke von zwei
verschiedenen Chromosomen miteinander verschmelzen. Damit kommt es auch
zur Verschemelzung von zwei Genen die normalerweise nichts miteinander
zu tun haben, zu einem Chimärengen.
Eine Vielzhal dieser Translokationen mit ihren Chimärengen werden bei
Leukämien gefunden. Häufig kommen die Translokationen (8;21) und
(15;17), sowie die t(9;22) vor. Die aus diesen Translokationen
abge-leiteten Genprodukte AML-1ETO, PML/RAR und BCR/ABL sind fir die
Entstehung der Leukämie verantwortllich.
Um diese Wirkung ausüben zu können, müssen sich diese Fusionsproteine in
den Blutvorläuferzellen zu sog. Makrokomplexen zusammenlagern. Diese
Makrokomplexe sind Strukturen, in denen die Leukämieinduzierenden
Fusionsproteine selbst als Oligomere vorliegen, die wieder an andere
Proteine gebunden sind. Wenn die Fähigkeit der Fusionsproteine
Makrokomplexe zu bilden, gehemmt wird, z. B durch die Inhibition der
Oligomerisierung, sind sie nicht mehr in der Lage Leukämie zu
induzieren.
Am Beispiel von BCR/ABL konnten wir ein neuartiges Therapieprinzip
entwickeln, das auf der Hemmung der Oligomerisierung mit Hilfe von
Peptiden, d. h. kleiner Aminosäuresequenzen basiert.
Das Problem dieses Ansatzes besteht darin, dass Peptide leider im Blut
sehr schnell abgebaut werden und deshalb die leukämischen Zellen nur
sehr schwer erreichen können.
Im Rahmen dieses Projektes soll über genetisch modifizierte
Knochmarkstromazellen, welche in die Lage versetzt werden, diese Peptide
zu produzieren und auszuschütten, versucht werden, die Peptide in die
unmittelbare Nähe der Zellen im Knochenmark zu bringen, von denen die
Leukämie ausgeht. Dazu soll die Eigenschaft der Stromazellen ausgenutzt
werden, eine Nische im Knochenmark zu bilden, in denen sich die
Leukämievorläuferzellen einnisten müssen, so dass ein enger Kontakt
gewährleistet ist, der eine parakrine
(direkt von Zelle zu Zelle) Übertragung der Peptide erlaubt, ohne sie
dem Abbau im Blut auszusetzen.
Solche modifizierten Stromazellen könnten in Zukunft in
Leukämiepatienten transplantiert werden und damit einen völlig neuen
Therapieansatz darstellen.
siehe auch:
http://www.nature.com/leu/journal/v23/n12/abs/leu2009194a.html
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3269486/
The effect of the dual Src/Abl kinase inhibitor AZD0530 on Philadelphia
positive leukaemia cell lines
nach oben
zur Projektseite
home
06/6
Wirkung von
Histonadeacetylase-Inhibitoren auf die dirrerentielle Genexpression in
hämatopoetischen Stammzellen
Frau Dr. med. Gesine Bug Klinik
für Kinderheilkunde III, Klinikum der
J.W.Goethe Universität
Frankfurt
Die Pluripotenz früher
hämatopoetischer Stammzellen (HSC) wird auf die offene Chromatinstruktur
dieser Zellen zurückgeführt, die die Transkription von Genen des
gesamten hämatopoetischen Ent-wicklungsprogramms erlaubt. In den letzten
Jahren wurde eine Reihe epigenetisch wirkender Substanzen entwickelt,
die die Genexpression durch reversible Veränderungen einzelner
Chromatinabschnitte durch Histondeacetylase (HDAC)-Inhibotoren
beeinflussen. Welche Veränderungen diese Chromatinmodellierung in
Stammzellen auslöst und welche stammzellregulatorischen Gene und
Signalwege von diesen Veränderungen betroffen sind, ist bisher nicht
untersucht.
Vorarbeiten:
wir konnten zeigen, daß der HDAC-Inhibitor Valproinsäure (VPA) sowohl
die Zellzyklusprogression und damit die Proliferation als auch die
Selbsterneuerung kurz- und langzeitpopulierender HSC verstärkt. Der in
früher klinischer Entwicklung befindliche HDAC-Inhibitor LAQ824 hemmt
ebenfalls die Differenzierung HSC, stimuliert aber im Gegensatz zu VPA
deren Proliferation nur wenig (siehe Abb.)
Vergleichbare Ergebnisse anderer Arbeitsgruppen mit dem HDAC-Inhibitor
Trichostatin A sprechen dafür, daß die Erhaltung und/oder Stimulation
funktionsfähiger HSC ein Klasseneffent der HDAC-Inhibitoren darstellt.
|
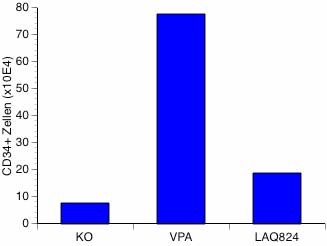
|
Abb.
LAQ824 hemmt die Differenzierung CD 34+ HSC bei
im Vergleich zu VPA geringerer Amplifikation
(Ergebnis nach 7-tätiger Suspensionskultur).
Bug G, Ritter M, Wassmann B,
Schoch C, Heinzel T, Schwarz K, Romanski A, Kramer OH, Kampfmann M,
Hoelzer D,
Neubauer A, Ruthardt M, Ottmann OG. Clinical trial of valproid acid and
all-trans retionic acid in patients with poor-risk acute myeloid
leikemia. Cancer. 2005 Dec 15;104(12):2717-25.
Bug G, Gul H, Schwarz K, Pfeifer H, Kampfmann M, Zheng X,
Beissert T, Boehrer S, Hoelzer D, Ottmann OG, Ruthardt M. Valproic acid
stimulantes proliferation and self-renewal of hematopoietic stem cells.
Cancer Res. 2005 Apr 1;65(7):419-26.
Romanski A, Bacic B, Bug G, Pfeifer H, Gul H, Remiszewski S, Hoelzer D,
Atadja P, Ruthardt M, Ottmann OG. Use of an novel histone deacetylase
inhibitor to
induce apoptosis in cell lines of acute lymphoblastic leukemia.
Haematologica 2004 Apr;89(4):419-26
Fragestellung:
Im Rahmen dieses Projektes soll die Wirkung verschiedener
HDAC-Inhibitoren auf die differentielle Genexpression früher HSC
untersucht werden. Dazu sollen Genexpressionsanalysen humaner CD 34-CD38-
HSC durchgeführt werden. Es wird eine Regulation von
Genen erwartet, die
1. die Erhaltung bzw. Differenzierung HSC oder
2. den Zellzyklus und damit die Proliferation HSC regulieren. Die
differenzelle Expression solcher
Gene soll auf RNA- und Proteinebene validiert werden.
Klinische Relevanz des Projektes:
Ältere Patienten mit AML haben trotz intensiver Chemotherapie
weiterhin eine sehr geringe Chance, von der Erkrankung geheilt zu
werden. Die Weiterentwicklung vielversprechender und gut verträglicher
molekularer Therapien wie der Differenzierungstherapie mit
HDAC-Inhibitoren ist deshalb dringend geboten und wird derzeit intensiv
geprüft. Die Rationale für den Einsatz von HDAC-Inhibitoren in der
AML-Behandlung ergibt sich aus ihrer differenzierungs- und
apoptoseinduzierenden Wirkung auf die Gesamtpopulation der AML-Blasten.
Die AML aber ist eine Stammzellerkrankung, die sich aus funktionell
unterschiedlichen Zellpopulationen zusammensetzt: der Masse nicht mehr
teilungsfähiger Blasten und dem kleinen Pool leukämischer Stamm-zellen,
die die AML erhalten. Kurative Therapieansätze müssen deshalb die
Stammzellen innerhalb der leukämischen Zellpopulation eradizieren, um
Rezidive zu verhindern. Aufgrund unserer Voruntersuchungen muß
befürchtet werden, daß HDAC-Inhibitoren auch leukämische Stammzellen
stimulieren, was zur Zeit weder bei der Therapieplanung noch bei
klinischen Begleituntersuchungen berücksichtigt wird.
Das Projekt soll zur Entwicklung gezielter, präferentiell gegen
AML-Stammzellen gerichteter Kombinations-therapien mit HDAC-Inhibitoren
beitragen.
siehe Veröffentlichung:
http://www.haematologica.org/content/92/4/542.article-info
Artikel in PDF
nach oben
zur Projektseite
home
06/7
Bildung eines
Pools, aus dem Kosten bestritten werden sollen, die bei
wissenschaftlichen Untersuchungen von Patienten und Stammzellspendern in
der Leukämietherapie entstehen und von Krankenkassen bzw. anderen
Kostenträger nicht übernommen werden
PD
Dr. H. Martin, Med. Klinik II
Klinikum der J.W. Goethe
Universität Frankfurt
06/8
Zellbiologische Charakterisierung von residualen Leukämiezellen bei
Kindern mit akuter lymphatischer Leukämie
Prof. Dr. med. W.-D.
Ludwig, Dr. L. Karawajew, Dr. R. Ratei
HELIOS Klinikum Berlin - Robert-Rössle-Klinik - Charité - Campus
Berlin-Buch Universitätsmedizin Berlin
Medizinische Klinik mit Schwerpunkt Hämatologie, Onkologie und
Tumorimmunologie
Zusammenfassung
Die
akute lymphatische Leukämie (ALL) ist die häufigste Krebserkrankung im
Kindesalter. Sie entsteht durch genetische Veränderungen, die
insbesondere Regulationsmechanismen der Zellteilung und des Zelltodes
betreffen. In der Folge kommt es zu einer starken Vermehrung der
unreifen Leukämiezellen, die gesunde Zellen im Knochenmark und Blut
verdrängen.
Bei der Therapie der kindlichen ALL sind synthetische Glucocorticoide,
die zu den Steroid-Hormonen gehören, von großer Bedeutung. Sie stellen
in der ersten Therapiewoche das Haupttherapeutikum dar. Bei etwa 60 %
der Patienten kann ihr Einsatz die Zahl der Leukämiezellen bereits
deutlich reduzieren. In den restlichen Fällen, wo die Wirkung weniger
effektiv ist, haben die Patienten später ein höheres Rückfall-Risiko und
benötigen in der Folge eine intensivere Chemotherapie.
Damit kommt den Leukämiezellen, die unter der Therapie persistieren
können und als minimale Resterkrankung (MRD) bezeichnet werden, eine
besondere prognostische Bedeutung zu. Das Ziel unseres Projektes
ist die zellbiologische Charakterisierung der MRD-Zellen, die wir zu
klinisch bedeutsamen Therapiezeitpunkten untersuchen.
Projektbeschreibung
Der Forschungsschwerpunkt unserer Arbeitsgruppe, der die Detektion und
Untersuchung von MRD bei kindlicher ALL zum Ziele hat, ist sowohl in das
Nationale Genomforschungsnetz (NGFN, gefördert von Bundesminister für
Bildung und Forschung) als auch in eine multizentrische Therapiestudie
bei Kindern (ALL-BFM) fest eingebunden.
In den letzten Jahren fokussierte sich unsere Arbeit auf die
Genexpressionsanalyse von MRD Zellen am Tag 8 der Induktionstherapie.
Die Ergebnisse unserer Untersuchungen zeigen, daß die resistenten
Leukämie-zellen, die die 1-wöchige Glucorticoid-Behandlung überlebt
haben, reifer erscheinen und eine geringere Zell-teilungsrate aufweisen
als die Masse der leukämischen Zellen zu Beginn der Therapie.
Dieser reifere, ruhende Zustand könnte zum einen die Resistenz der
Leukämiezellen erklären, und zum anderen Auswirkungen auf den weiteren
Verlauf der Chemotherapie haben. Es konnten auch Kandidaten-gene, die an
der Entstehung von Resistenz gegenüber der Chemotherapie beteiligt sein
könnten, identifiziert werden. Dazu gehören molekulare Faktoren, die
maßgeblich an der Regulation des Zelltodes (BCL-2, clAP2) und an der
B-Zell-spezifischen Signaltransduktion (pre-B-Zellrepeptor) beteiligt
sind. Außerdem haben wir bestimmte Rezeptormoleküle, deren Expression in
leukämischen Zellen bisher nicht beschrieben wurde (CD11b, IFNgammaR),
an der Zelloberfläche der persistierenden Leukämiezellen entdecken
können. Im weiteren Verlauf unseres Projektes sollen, auf den bisherigen
Erkenntnissen aufbauend, die zellbiologi-schen und molekularen
Veränderungen leukämisch Zellen zu späteren Therapiezeitpunkten (Tag 15,
Tag 33) untersucht werden. Hierzu werden die leukämischen Blasten
zunächst im Rahmen der MRD-Diagnostik immunphänotypisch charakterisiert.
Das dazu verwendete Protokoll ist von der internationalen MRD
Studien-gruppe entwickelt worden, die zum Ziel hat, die MRD Detektion
mittels multiparametrischer Durchfluß-zytometrie hinsichtlich
Sensivität, Durchführbarkeit und prognostischer Relevanz prospektiv zu
evaluieren.
Die anschließende Isolierung der Blasten erfolgt mit Hilfe eines
Zellsortierers. Die für die Genexpressions-analyse von residualen
Leukämiezellen mittels Mikroarraytechnik notwendigen methodischen
Schritte, einschließlich mRNA Isolierung und cRNA Amplifizierung aus
einer kleinen Blastenzahl sowie Hybridisierung von cRNA Proben mit
Affymetrix HG U133A Oligonukleotid-Chips wurden bereits etablliert.
Die Kandidaten für Wirk- und Resistenzmechanismen der
Induktionstherapie, die aus der zellbiologischen Charakterisierung der
MRD-Zellen hervorgegangen sind, sollen anschließend in in vitro-Zellsystemen
(primäres Tumormaterial und Zelllinien) validiert und auf molekularer
Ebene weiter untersucht werden.
nach oben zur Projektseite home
06/9
Einfluß von Zelltherapien auf die
Erholung des Immunsystems nach Stammzelltransplantationen bei Kindern
und Erwachsenen mit Leukämien
Frau Dr.rer. nat. Ulrike Köhl,
Leiterin
des Labors für Stammzelltranplantation und Immuntherapie der
Universitätskinderklinik Ffm.,
PD Dr. Hans Martin
Leiter der KMT-Einheit, Oberarzt der Inneren Medizin, Med. Klinik III,
Abt. Hämatologie und Onkologie – Klinikum der J.W. Goethe Universität
Frankfurt am Main
Akute Leukämien kommen in allen Altersstufen
vor. Im Kindesalter treten vorwiegend akute lymphatische Leukämien (ALL)
auf, während Erwachsene überwiegend an akuten myeloischen Leukämien
(AML) erkranken.
Zur Behandlung von Hochrisikopatienten mit
akuten Leukämien und bei Patienten mit einem Rückfall der Leukämie
bietet die Hochdosischemotherapie mit und ohne Ganzkörperbestrahlung und
anschließender Stammzelltransplantation eine Möglichkeit, die
Heilungschancen im Vergleich zur konventionellen Chemo-therapie zu
erhöhen. Bei der
Stammzelltransplantation erhält der Patient Stammzellen eines teilweise
oder sogar völlig passenden Fremd- oder Familienspenders.
Die verschiedenen patienteneigenen Zellen des Immunsytems, die durch die Therapie vor Stammzelltrans-plantation stark vermindert oder völlig ausgelöscht wurden, regenerieren nach Transplantation aus den frisch transplantierten Stammzellen des Spenders. Dabei erholen sich die verschiedenen Immunzell-Populationen qualitativ und quantitativ unterschiedlich schnell. Patienten mit rascher Erholung des Immunsytems erreichen meist innerhalb des ersten halben Jahres nach Transplantation eine gute Abwehr gegen Infektio-nen und erleiden selten einen Rückfall ihrer Erkrankung (Koehl 2007). Bei Patienten mit schlechter Regeneration des Immunsytems, die oft von schweren Infektionen und erneutem Auftreten der Leukämie betroffen sind, wird versucht durch zusätzliche Therapien mit Immunzellen des Spenders die Zellerholung zu fördern (Koehl 2004+2005).
Ziel des vorliegenden Antrags ist es daher, zunächst in einer bizentrischen Pilotstudie bei 10 pädiatrischen und 10 internistischen Patienten mit akuter Leukämie die Erholung der verschiedenen Immunzellen des Immunsystems und deren Funktion engmaschig zu untersuchen. Dank der Unterstützung durch die Gutermuth-Stiftung konnten immunologische Messmethoden zur Bestimmung der verschiedenen Immunzellen sowie ihrer Funktionalität, aufgebaut werden, die nun auch für eingeschickte Proben zur Verfügung stehen. In einem Jahr soll mit Hilfe der ersten 20 Patienten die Logistik für diese bizentrische Studie und eine Datenbank für die multivariaten Analysen etabliert werden. Anschließend ist die Aufnahme von 150 Patienten (in 5 Jahren) zur Untersuchung der altersabhängigen Immunrekonstitution und dem Einfluss der Zelltherapien auf das Immunsystem geplant. Die Studie soll entscheidend dazu beitragen, mit einer schnellen und kostengünstigen Diagnostik „Hochrisikopatienten“ zu ermitteln, um diese rechtzeitig nach Transplantation mit einer zusätzlichen Zelltherapie behandeln zu können. In einer statistischen Analyse soll die Zellerholung hinsichtlich Überleben, Infektion, Rückfall der Erkrankung, zusätzliche Zelltherapiebehandlung sowie Art der Transplantatquelle ausgewertet werden.
Forschungsergebnis:
Nach Stammzelltransplantation (SZT) sind die Patienten durch Viren sehr
gefährdet, z.B. durch die Reaktivierung mit dem Cytomegalovirus (CMV).
Wir konnten eine Studie zur Regeneration CMV-spezifischer T-Zellen
(CMV-CTL) nach SZT bei 278 Patienten mit Leukämien abschließen.
Patienten mit nur 1 CMV-CTL/μl im peripheren Blut zwischen Tag +50 und
+75 nach SZT hatten einen signifikant höheren Schutz vor einer
CMV-Reaktivierung gegenüber Patienten, die keine CMV-CTLs entwickelten
(Borchers PlosOne 2012). Um Patienten, die keine eigenen CMV-CTLs
entwickeln und nicht mehr auf antivirale Therapien ansprechen, mit
CMV-CTLs zu behandeln, konnten wir erfolgreich die Herstellung dieser
virusspezifischen T-Zellen auf ein vollautomatisches Gerät bringen
(Priesner et al. Frontiers Immunol 2016).
In weiteren Untersuchungen konnten wir die Bedeutung von Dendritischen Zellen auf die Immunrekonstitution nach SZT zeigen. Dazu wurden zunächst Normwerte zu Dendritischen Zellen im Peripheren Blut von gesunden Kindern erhoben, die für einen kleinen chirugischen Eingriff aufgenommen wurden (Heinze et al. Scand Immunol 2013).
siehe Veröffentlichung in Bone Marrow Transplantation (2010) 45, 613-621
nach oben zur Projektseite home
Vergleichende molekulargenetische Analyse von CD 34+ mit unselektierten hämatopoetischen Zellen aus dem Knochenmark von Patienten mit myelodysplastischem Syndrom (MDS)
Prof. Dr. Wolf-Karsten Hofmann, Med. Klinik III, Hämatologie, Onkologie, Transfusionsmedizin, Campus Benjamin Franklin, Charité – Universitätsmedizin Berlin
Es gibt zahlreiche Hinweise dafür, dass die
Entstehung eines MDS auf die Anhäufung genetischer Schäden in
blutbildenden Zellen des Knochenmarks zurückzuführen ist. Über die Art
dieser Schäden ist bisher jedoch nur wenig bekannt.
Kürzlich wurde mit der Einführung von „500K SNP mapping arrays“ eine
sehr robuste Technologie auf der Basis von „DNA-Chips“ entwickelt, die
erstmals eine detaillierte Analyse dieser genetischen Veränderungen
erlaubt. Mit diesen DNA-Chips können kleinste (bisher nicht sichtbare)
Veränderungen am genetischen Material in den Blutzellen dargestellt
werden.
Bisher unklar ist auch, ob die genetischen Veränderungen ausschließlich blutbildende Vorläuferzellen von MDS-Patienten betreffen, oder ob es in den Knochenmarkzellen (die auch ausgereifte Zellen enthalten) gleichfalls signifikante Veränderungen gibt. Weiterhin unklar ist, wie sehr die genetischen Profile der einzelnen Zellarten miteinander im Zusam-menhang stehen.
Das vorliegende Projekt wird erstmalig unter Anwendung der hochauflösenden DNA-Chips diese wichtige vergleichende Analyse von blutbildenden Vorläuferzellen und unselektierten Knochenmarkzellen beim MDS ermöglichen.
siehe auch:
Veröffentlichung in Experimental Hematology
http://www.exphem.org/article/S0301-472X(08)00487-6/abstract
nach oben
zur Projektseite
home
Epigenetische Regulation des Notch-Signalwegs in normalen und leukämischen Stammzellen
Frau Dr. med. Gesine Bug Oberärztin, Med. Klinik II, Abt. Hämatologie/Onkologie, J.W.Goethe Universität
60590 Frankfurt, Theodor-Stern-Kai 7
Ältere Patienten mit akuten Leukämien haben trotz intensiver
Chemotherapie weiterhin eine sehr geringe Chance, von der Erkrankung
geheilt zu werden. Eine Verbesserung der Leukämietherapie ist durch den
Einsatz neuer molekularer Medikamente zu erwarten, die spezifisch auf
Leukämiezellen wirken, ohne die normale Blutbildung (Hämatopoese) zu
zerstören. Eine vielversprechende Substanzgruppe stellen
Histondeacetylase-Inhibitoren (HDACi) dar.
Die gesamte Hämatopoese geht auf eine kleine Population blutbildender Stammzellen zurück, die den Körper ein Leben lang mit reifen, funktionstüchtigen Blutzellen versorgen. Die akute myeloische Leukämie (AML) ist eine Stammzellerkrankung, bei der es durch unkontrollierte Vermehrung und gestörte Ausreifung bösartiger (leukämischer) Stamm-zellen zu einer Anhäufung unreifer Leukämiezellen (Blasten) im Knochen-mark kommt. In Laborversuchen bewirken HDACi eine Umwandlung der Blasten in reife Blutzellen, die sich nicht mehr teilen können und zugrunde gehen. Die ersten klinischen Erfahrungen zeigen, daß eine Behand-lung mit HDACi möglich, aber bei den meisten AML-Patienten nicht ausreichend wirksam ist.
Um eine Heilung zu erzielen, muß eine AML-Therapie nicht nur Blasten, sondern auch die leukämischen Stammzellen zerstören. Wir haben deshalb den Effekt der HDACi zunächst auf normale blutbildende Stammzellen analysiert und eine unerwartete Beobachtung gemacht: HDACi verzögern die Stammzellreifung und können sogar die Vermehrung der Stammzellen unterstützen. Die Wirkung der HDACi auf leukämische Stammzellen ist bisher nicht untersucht. Es ist aber denkbar, daß HDACi zwar Blasten in den Zelltod treiben, aber die für den Verlauf der Erkrankung entscheidenden leukämischen Stammzellen nicht vernichten.
Ziel unseres Projektes ist es, die Bedeutung des Notch-Signalwegs für die Wirkung des HDACi LAQ824 auf normale und leukämische Stammzellen zu untersuchen. Dies ist klinisch von großer Bedeutung, weil eine Hemmung des Notch-Signalwegs durch mTOR-Inhibitoren wie das zur Immunsuppression zugelassene Rapamycin (Sirolimus), beschrieben ist. Eine Verbesserung der antileukämischen Wirkung der HDACi durch Kombination mit Antagonisten des Notch-Signalwegs liegt also nahe und soll im leukämischen Maus-modell geprüft werden.
nach oben zur Projektseite home
07/2
Klinische
Relevanz der funktionalen Interaktionen zwischen erhöhter genomischer
Instabilität, deregulierter DNA-Schädigung und defekter Apoptose bei
myelodysplastischem Syndrom und akuter myeloischer Leukämie
Frau PD Dr. Simone Boehrer,
Institut Gustave Roussy, Villejuif, Frankreich, in Kooperation mit
Herrn
PD Dr. Ruthardt,Med.
Klinik II / Hämatologie, Klinikum der J.W. Goethe Universität Frankfurt
Bei myelodysplastischen Syndromen
(MDS) handelt es sich um eine heterogene Gruppe bösartiger Erkrankungen,
die bevorzugt im erhöhten Lebensalter auftreten, und die mit einer
erheblich unterschiedlichen Wahrscheinlichkeit - über einen Zeitraum von
mehreren Monaten bis wenigen Jahren - in eine akute myeloische Leukämie
(AML) übergehen. Die klinischen Faktoren, die diese Wahrscheinlichkeit –
und damit auch die Überlebenszeit der einzelnen Patienten – bestimmen,
sind bereits gut etabliert. Die tumorbiologischen Voraussetzungen, die
dieser Evolution von eher indolenten Stadien zu zunächst aggressiveren
Verlaufsformen des MDS und schließlich der (aufgrund des höheren
Lebensalters/be-stehender Komorbidität und unzureichender
Therapiealternativen) fast ausnahmlos letal verlaufenden AML
zugrundeliegen, sind noch unzureichend bekannt.
Anhand von kürzlich erschienenen Publikationen wurden neure, wegweisende
Schlussfolgerungen hinsichtlich der Mechanismen, welche die Entstehung
und Propagation von soliden Tumoren ermöglichen, gezogen: diese
Untersuchungen zeigen, daß es während der “schrittweisen” Entwicklung
solider Tumoren vom “noch normalen, aber bereits geschädigten
Zellverband” bis zum metastasierungsfähigen Malignom zeitgleich zu einer
schrittweisen, funktionellen Einschränkung der Signalwege kommt, die im
gesunden Zellverband aktiviert werden können (DNA-damage response).
Diese Aktivierung der Signalwege, die analog in MDS und AML Zellen
aktiviert werden können, führt dann dazu, dass geschädigte Zellen
(insbesondere mit erhöhter genomischer Instabilität, das heißt solche
Zellen, die bei Teilung eine fehlerhafte DNA an ihre Tochterzellen
weitergeben können) dem programmierten Zelltod unterworfen werden. An
soliden Tumoren konnte somit schlüssig belegt werden, welche Moleküle
innerhalb der DNA-damage response Signalwege während der Tumorevolution
inaktiviert werden und somit die Etablierung maligner Zellverbände
ermöglichen.
Unser Projektvorschlag soll die Hypothese überprüfen, dass auch während
der Entwicklung vom Niedrigrisiko-, über das Hochrisiko-MDS bis zur AML
eine solche schrittweise funktionelle Inaktivierung der Signalwege
stattfindet, die schließlich zur Entstehung genomisch instabiler
Zellverbände führt, die sich - zunehmend erfolgreich - dem
programmierten Zelltod (auch dem durch Chemotherapien induzierten
programmierten Zelltod) entziehen können.
Um diese Hypothese überprüfen zu können, bedienen wir uns im Labor
bereits vorhandener Zelllinienmodelle, welche unterschiedliche
Subgruppen des MDS und der AML repräsentieren.
Diese werden sowohl vor als auch nach einem DNA-schädigenden Stimulus (g-Bestrahlung) hinsichtlich der Funktionalitaet ihrer Signalwege untersucht. Dabei werden – analog den Ergebnissen an soliden Tumoren – molekulare Ereignisse identifiziert, welche auf dem Weg von der praeneoplastischen Laesion bis zur Leukämie eine Schlüsselstellung einnehmen. Im zweiten Teil des Projects – und insbesondere für diesen wird die Förderung durch die Gutermuth-Stiftung beantragt – soll die klinische Relevanz dieser Ergebnisse verifiziert werden. Zu diesem Zweck wird histologisches Material (Knochenmark) von Patienten mit den unterschiedlichen Verlaufsformen des MDS/der AML hinsichtlich der Expression dieser “molekularen Schlüsselmoleküle” untersucht werden. Die Ergebnisse lassen nicht nur erwarten, dass wesentliche Rückschluesse zur Pathogenese dieser Erkrankungsgruppe gezogen werden können, sondern ermöglichen wahrscheinlich auch die Identifikation zukünftiger therapeutischer Angriffspunkte.
nach oben zur Projektseite home
07/3
Bedeutung von microRNA für das Therapieansprechen und die
Resistenz bei akuten Leukämien
Prof. Dr. W.-D. Ludwig, Dr. L.
Karawajew , Dr. R. Ratei, P. Rhein, Universitätsmedizin Berlin Charitè
- Campus Buch, Robert-Rössle-Klinik mit Schwerpunkt Hämatologie,
Onkologie u. Tumorimmunologie
Die Entschlüsselung der Gensätze von
verschiedenen tierischen und pflanzlichen Arten führte vor kurzem zur
Entdeckung einer neuen Klasse regulatorischer Gene, deren Produkte als
microRNAs (miRNA) bezeich-net werden.
Gene produzieren im Allgemeinen Proteine, die spezifische Funktionen in
der Zelle einnehmen. Im Vergleich zu den Proteinen stellen miRNAs sehr
viel kleinere Moleküle dar, deren Aufgabe ist es, die Bildung anderer
Genprodukte zu verhindern. Auf diesem Weg haben sie entscheidenden
Einfluss auf die Ausführung von zellulären Prozessen.
Nach neuen Studien könnten microRNAs eine
wichtige Rolle in der Entwicklung, bei Krebs und beim Stoffwechsel
spielen. Es wurde unter anderem gezeigt, dass sie an der
Regulation der Blutbildung und des Zelltodes beteiligt sind und damit
auch für die Entstehung von Krebserkrankungen des Blutes (Leukämien) in
Frage kommen.
In unserem Projekt stellen wir die Frage über die Bedeutung der miRNAs
für das Ansprechen und die Resistenz von Leukämiezellen bei der
therapeutischen Behandlung.
Die Untersuchungen werden sich vor allem auf akute Leukämien im
Kindesalter fokussieren, und sollen neue, klinisch relevante Einblicke
in die bislang noch unzureichend aufgeklärten Mechanismen der
Therapieresistenz bei diesen Krankheiten ermöglichen.
nach oben zur Projektseite home
07/4
Funktionelle
molekularbiologische Charakterisierung von Calreticulin als
MDS-spezifisches Targetgen
Prof. Dr. Wolf-Karsten Hofmann, Med.
Klinik III, Hämatologie, Onkologie, Transfusionsmedizin,
Campus Benjamin Franklin, Charité – Universitätsmedizin Berlin
Die Nutzung molikulargnetischer Methoden zur Analyse von Blutstammzellen
von Patienten mit myelodys-plastischem Syndrom (MDS) haben zu einer
Reihe von Enedeckuongen geführt, die Hinweise auf verschie-dene
menschliche Gene geben, die an der Entstehung dieser Krankheit beteiligt
sein könnten.
In unseren Vorarbeiten haben wir unter anderem erstmalig uner Anwenduong
der hochauflösenden DNA-Chips MDS-Stammzellen untersucht und eine Reihe
von veränderten Genene gefunden.
Das dabei identfizierte Gen Calreticuilin (CALR) ist in Stammzellen von
MDS-Patienten praktisch abgeschal-tet, obwohl es in gesunden Zellen eine
wichtige Funktion beim Wachstum und bei der Ausreifung von Blut-zellen
hat. Das ist der Hinweis darauf, dass dieses Gen wahrscheinlich
ursächlich bei der Entstehung eines MDS beteiligt ist.
Im vorliegenden Projekt
sollen die biologischen Eigenschaften und die Funktion deses Gens genau
unter-sucht werden. Durch "An - und Abschalt-Experimente" in Blutzellen
soll es gelingen, Erkenntniss über die Störung deses Gens beim MDS zu
erhalten und damit vielleicht einen wichtigen Faktor für die Grundlagen
dieser Krankheit aufzudecken.
nach oben
zur Projektseite
home
07/5
Charakterisierung der Chromosomenbruchpunktregionen bei
Major breakpoint
BCR-ABL-positiver ALL und CML
Dr. med.
Dr. rer. nat. Thomas Burmeister, Charité Universitätsmedizin Berlin
Campus Benjamin Franklin ,Medizinische Klinik III, Hindenburgdamm 30
12200 Berlin
Hintergrund: Fast alle Patienten mit chronischer myeloischer Leukämie (CML) und etwa 25% der erwach-senen Patienten mit akuter lymphatischer Leukämie (ALL) zeigen eine spezifische genetische Veränderung, das so genannte Philadelphia-Chromosom. Auf molekularer Ebene kommt es dabei zu einer Chromosomen-translokation t(9;22)(q34;q11) und zur Fusion von zwei Genen, „BCR“ (auf Chromosom 22) und „ABL“ (auf Chromosom 9). Dadurch wird ein neuartiges Fusionsgen „BCR-ABL“ gebildet, das als Onkogen (krebsaus-lösendes Gen) wirkt und als eigentliche Ursache der Erkrankung gilt.
Fragestellung und Zielsetzung: Die Ursachen des Chromosomenbruchs sind bisher nicht verstanden. Bestimmte Chromosomenbruchpunktregionen („major breakpoint“, „Minor breakpoint“) sind bekannt, jedoch nicht die genaue Lokalisation der Chromosomenbruchpunkte. Ziel des vorliegenden Projektes ist es, bei einer größeren Zahl von Patienten mit CML und BCR-ABL-positiver ALL die entsprechenden Chromosomen-bruchpunkte in der „major breakpoint region“ zu ermitteln. Dadurch lässt sich ein vertieftes molekulares Verständnis dieser genetischen Aberration und damit der Krankheitsursache gewinnen. Möglicherweise lassen sich auch Risikofaktoren für das Auftreten der Chromosomentranslokation bestimmen, die eine bessere Prävention ermöglichen könnten.
Abbildung: Chromosomenbruchpunktregionen in den Genen BCR und ABL. 70% der BCR-ABL-positiven ALL-Patienten zeigen einen Bruchpunkt in der minor breakpoint region (m-bcr) und 30% in der Major breakpoint region (M-bcr). Fast alle CML-Patienten haben einen Chromosomenbruch in der M-bcr. Im Bereich von ABL liegt der Bruchpunkt zwischen Exon Ib und Exon 2.
Ergebnisse:
Im Rahmen
des Forschungsprojektes wurde eine Methode entwickelt, mit der sich mit
einem verhältnis-mäßig geringen Aufwand die Position des
Chromosomenbruchs der Philadelphia-Chromosomentranslokation ermitteln
läßt.
Das ist eine nichttriviale Aufgabe, da die beiden an der Translokation
beteiligten Chromosomen 9 und 22 mehrere 100 Millionen Nukleotidbasen
groß sind.
Mit der entwickelten Methode, einer sogenannten inversen PCR, lässt sich
der Chromosomenbruchpunkt auf eine Nukleotidbase genau bestimmen.
Unter Anwendung dieser Methode gelang die Charakterisierung der
Chromosomenbruchpunkte von 64 Patientien mit
Philadelphia-Translokation (CML
oder ALL). Dadurch wurden vertiefte
Einsichten in den molekularen Mechanismus dieser Veränderung gewonnen.
Außerdem ist die entwicklete Methode auch zur Charakterisierung
ähnlicher Veränderungen geeignet.
Veröffentlicht unter:
Burmeister T, Gröger D, Kuhn A, Hoelzer D,
Thiel E, Reinhardt R: Fine structure of translocation
breakpoints within the major breakpoint region in
BCR-ABL1-positive leukemias. DNA Repair (2011),
doi:10.1016/j.dnarep.2011.08.010
nach oben zur Projektseite home
07/6
Quantitative Analyse der DNA-Methylierung von differenzierenden
erythropoetischen Zellen von Patienten mit Myelodysplastischem Syndrom
Prof. Dr. Wolf-Karsten Hofmann,
Med. Klinik III, Hämatologie, Onkologie, Transfusionsmedizin,Campus
Benjamin Franklin, Charité – Universitätsmedizin Berlin
Beim Myelodysplastischen Syndrom
(MDS) liegt eine schwere Störung und Entartung der Blutzellen vor.
In den letzten Jahren hat man erkannt, daß das gestörte Wachstum dieser
Zellen ursächlich mit Veränder-ungen im Bereich der Chromosomen oder
aber einzelner Gene verknüpft ist. Dies ist auch die Ursache dafür, daß
es später zur Entwicklung der bösartigen akuten myeloischen Leukämie
kommt.
Im Bereich der Gen-Informationen,
die in der menschlichen DNA gespeichert sind, gibt es beim MDS
ver-schiedene Veränderungen wie den Verlust von DNA-Abschnitten
(Deletionen), Veränderungen des genetischen „Codes“ in den MDS-Zellen
(Mutationen), Vervielfachungen von DNA-Abschnitten (zum Beispiel
Verdoppelungen – Duplikationen) und andere.
Außerdem hat man durch systematische Untersuchungen herausgefunden, daß
einige Abschnitte der DNA auf eine bestimmte Art und Weise chemisch
modifiziert („methyliert“) sind.
Im Zusammenhang mit einem Projekt,
welches bereits von der Gutermuth-Stiftung gefördert wurde, konnten
solche chemisch veränderten Abschnitte auf der DNA von MDS-Zellen
gefunden werden.
Mit einer sehr neuen experimentellen Technik (sogenanntes
„Pyrosequencing“) kann es gelingen, die chemisch veränderten
DNA-Abschnitte nicht nur nachzuweisen, sondern auch die Schwere der
Veränderung (Quantifizierung) zu bestimmen.
Mit Hilfe dieser Technik sollen nun – in konsequenter Fortsetzung der
bereits durchgeführten Arbeiten, die Quantifizierung der
DNA-Methylierung in Blutzellen von MDS-Patienten vorgenommen
werden
nach oben
zur Projektseite
home
08/1
Epigenetische Stammzellsignatur durch die APL-assoziierten
Fusionsproteine PML/RAR a und PLZF/RAR a
PD.
Dr./Univ. Perugia Martin Ruthardt, Klinikum der J.W. Goethe-Universität,
Med. Klinik III, Hämatologie, Theodor-Stern-Kai 7 60596 Frankfurt,
Die Epigenetik untersucht über die Phasen der
Zellteilung hinaus vererbbare Veränderungen der Genfunktion, die nicht
mit Veränderungen der Erbinformation in der DNA-Sequenz der Chromosomen
erklärt werden können.
Epigenetischen Aberrationen (Abweichungen) wird eine entscheidende
Bedeutung für die Tumorgenese zugeschrieben. Die hauptsächlichen
epigenetischen Veränderungen sind Modifikationen an den strukturellen
Untereinheiten der Chromosomen, den Nukleosomen, deren Funktion über
Zustandsänderungen ihrer Bausteine, der Histone, um die die
Erbinformation der DNA „herumgewickelt“ ist, gesteuert werden kann.
Weitere bedeutende epigenetische Veränderungen spielen sich direkt an
der DNA ab, wie die Methylierung ( chemische Abänderung ) der DNA.
Man geht davon aus, daß es eine Sequenz epigenetische Aberration,
gefolgt von DNA-Methylierung wiederum gefolgt von Mutationen in der
Erbinformation im Rahmen der Leukämogenese gibt. Deshalb wird vermutet,
daß ein direkter Zusammenhang zwischen aberranter Epigenetik und
Mutationen existiert, die den veränderten bösartigen Zustand der Zelle
festschreiben. Das würde bedeuten, daß es einen Zeitraum in der
Krankheitsentstehung gibt, in dem die Veränderungen der bösartigen
Zellen, und zwar die epigenetischen Veränderungen, rückgängig gemacht
werden können, da man mit Medikamenten, den epigenetischen Zustand von
Zellen beeinflussen kann, die aber natürlich nichts mehr nützen, wenn
Mutationen aufgetreten sind, die den bösartigen Zustand der Zellen
festschreiben.
Große Schwierigkeiten bei den Untersuchungen zum Einfluß von
epigenetischen Aberrationen durch Leukämie-induzierende Ereignisse wie
z.B. Chromosomen-translokationen und ihre Fusionsproteine, macht das
„epigenetische Hintergrund-rauschen“ der untersuchten Zellen; d.h. es
gibt sehr viele epigenetische Verände-rungen, die mit der Leukämogenese
sicher nichts zu tun haben, sondern einfach auf den Funktionszustand,
d.h. Differenzierungsgrad, Gewebezugehörigkeit usw., zurückgeführt
werden können.
Diese Fragestellungen sollen in einer Sequenz von Zellmodellen
untersucht werden, die durch eine zunehmende epigenetische
Determinierung charakterisiert sind. Ausgehend von embryonalen
Stammzellen der Maus, die den niedrigsten Grad der epigenetischen
Determinierung und damit das geringste unerwünschte „epigenetische
Hintergrund-rauschen“ aller möglichen Zellmodelle aufweisen dürften,
weiter über sog. embryonale Karzinomzellen zu primitiven adulten
hämopoetische Stammzellen wird am Beispiel der Leukämie-assoziierten
Fusionsproteine PML/RAR und PLZF/RAR der Einfluß auf einzelne
epigenetische Ereignisse in diesen Zellen untersucht werden.
Im Rahmen dieses Projekts soll geklärt werden, welche epigenetischen Veränderungen tatsächlich für die Leukämogenese eine Rolle spielen und deshalb in einer Frühphase der Erkrankung möglicherweise therapeutisch angegriffen werden sollen.
siehe Veröffentlichung http://cancerres.aacrjournals.org/content/65/14/6080.full.pdf
nach oben zur Projektseite home
Funktionelle molekularbiologische Charakterisierung von Alcam, ALPHA CATENIN und CALRETICULIN als MDS-spezifische Targetgene
Prof. Dr. Wolf-Karsten Hofmann, Med. Klinik III, Hämatologie, Onkologie, Transfusionsmedizin,Campus Benjamin Franklin, Charité – Universitätsmedizin Berlin
In unseren Vorarbeiten haben wir
durch die Nutzung molekulargenetischer Methoden zur Analyse von
Blut-stammzellen von Patienten mit myelodysplastischem Syndrom (MDS)
erstmalig unter Anwendung der hoch-auflösenden DNA-Chips MDS-Stammzellen
untersucht und eine Reihe von veränderten Genen gefunden.
Zunächst wurde begonnen, das dabei identifizierte Gen Calreticulin
(CALR), welches in Stammzellen von MDS-Patienten praktisch abgeschaltet
ist, obwohl es beim Gesunden eine wichtige Funktion beim Wachs-tum und
bei der Ausreifung von Blutzellen hat, funktionell zu charakterisieren.
In diesem Zusammenhang durchgeführte weitere molekulargenetische
Analysen zeigten eine Störung der Gene ALCAM und alpha-Catenin – beides
wichtige Gene für das normale Zellwachstum
Im vorliegenden Projekt sollen in Fortführung der Arbeiten zu Calreticulin die biologischen Eigenschaften und die Funktion dieser neu identifizierten Gene genau untersucht werden. Erneut sollen „An- und Abschalt-Experimente“ in Blutzellen dazu beitragen, Erkenntnisse über die Störung dieser Gene beim MDS zu erhalten.
nach oben zur Projektseite home
08/3
Assoziation von genetischen Polymorphismen
des angeborenen Immunsystems mit dem Verlauf der Immunrekonstitution
nach Stammzelltransplantation und dem Risiko für infektiöse
Komplikationen sowie der Graft-versus-Host-Erkrankung
Frau PD
Dr. Ulrike Köhl Klinikum der J.W.
Goethe-Universität, Zentrum für
Kinder- und Jugendmedizin, Klinik für Kinderheilkunde III, Pädiatrische
Hämatologie, Onkologie und Hämostaseologie, Labor für
Stamm-zelltransplantation und Immuntherapien, Theodor-Stern-Kai 7, 60596
Frankfurt am Main
Für Hochrisikopatienten mit akuten Leukämien
konnte durch die allogene Stammzell-transplantation (SZT) die
Heilungsrate im Vergleich zur konventionellen Chemotherapie deutlich
erhöht werden. Trotzdem sterben weiterhin etliche Patienten nach SZT an
einem Rezidiv ihrer Erkrankung, an einer schweren Infektion oder an
einer Graft-versus-Host-Erkrankung (GvHD). Dabei sind eine langsame
Immunrekonstitution und bestimmte genetische Polymorphismen (Auftreten
von Genvarianten) mit einem hohen Risiko für eine Infektion oder eine
GvHD verknüpft.
Im Rahmen dieses Projektes soll überprüft werden, inwieweit Polymorphismen von Molekülen des angebore-nen Immunsystems den Verlauf der Immunrekonstitution und das Risiko für infektiöse Komplikationen oder für eine GvHD nach SZT beeinflussen. Dazu soll mittels Real-time PCR eine Genotypisierung der Spender und der Empfänger (n=50) hinsichtlich verschiedener Polymorphismen bestimmt werden. Anschließend werden die Daten hinsichtlich Überleben, Auftreten einer Infektion oder einer GvHD nch SZT ausgewertet.
siehe Veröffentlichung:
PLoS One.
2012;7/(12):e50248.doi:10.1371/journal.pone.0050248. Epub 2012 Dec 13.
Erratum in: PLoS One. 2013;8(4). doi:
10.1371/annotation/43e9b84c-fbe3-4b39-88c8-1cde34bOafea.
nach oben zur Projektseite home
08/4
Quantitative Analyse der DNA-Methylierung
von Zellzyklusregulierenden Genen sowie von "Genomic-Clock-Markern" in
der menschlichen Hämatopoese
Prof. Dr. med. Wolf-K. Hofmann
Medizinische Klinik III Campus
Benjamin Franklin Charité - Universitätsmedizin Hindenburgdamm 30,
2203 Berlin
Hintergrund
Das myelodysplastische Syndrom (MDS)
ist eine Modellerkrankung der Hämatopoese. Am Beispiel des MDS konnte
bisher durch verschiedene zellbiologische und molekulargenetische
Methoden gezeigt werden, daß für die leukämische Transformation der
hämatopoetischen Stammzelle die Kumulation von endogenen und exogenen
Ereignissen erforderlich ist. In Vorarbeiten zur Genexpression und zur
DNA-Methylierung in adulten hämatopoetischen Stammzellen (sowohl normale
Zellen als auch Zellen von Patienten mit MDS) konnte gezeigt werden, daß
es MDS-spezifische Veränderungen der DNA-Methylierung für einzelne Gene
(z. B. p15INK4b) gibt. Auf diese Daten stützt sich auch der
klinische Einsatz von demethylierenden Substanzen wie 5-Aza-Cytidine
oder 5-Deoxy-2-Cytidine. Erkenntnisse über quantitative
DNA-Methylierungsmuster von zellzyklusregulierenden Genen während der
normalen und krankhaften Entwick-lung der hämatopoetischen Stammzelle in
allen drei Linien der Hämatopoese sind bis jetzt nicht verfügbar. Diese
systematische und quantitative Untersuchung der differenzierenden
Hämatopoese ist im vorliegenden Projekt geplant. Neueste Erkenntnisse
geben Hinweise darauf, daß nicht nur der Ort der DNA-Methylierung,
sondern auch die Ausprägung (Anzahl der methylierten Abschnitte) für die
Regulation der Genexpression von Bedeutung ist. Es konnte gezeigt
werden, daß durch quantitative DNA-Methylierungsanalyse das mitotische
Zellalter (also letztendlich die Anzahl der bisher stattgefundenen
Zellteilungen) bestimmt werden kann. Damit hat man eine bisher nicht
verfügbare Möglichkeit, Störungen in der Proliferation und
Differenzierung von hämatopoetischen Zellen zu untersuchen.
Fragestellung und Ziele
Ziel des Projektes ist es, das mitotische Zellalter in hämatopoetischen
Zellen von Patienten mit MDS und mit Akuter Leukämie zu untersuchen. Als
Vergleich werden analoge Analysen an hämatopoetischen Zellen von
gesunden Probanden durchgeführt. Zur quantitativen Bestimmung der
DNA-Methylierung wird ein völlig neues Verfahren („Pyrosequencing“)
angewandt. Durch die Abschätzung des Anteils von Zellen mit einem hohen
mitotischen Alter (typisch für pathophysiologisch erhöhte Zellteilung)
soll es gelingen, Aufschluß über den Ursprung dieser malignen
hämatologischen Systemerkrankungen zu erhalten. Insbesondere soll die
Frage nach der Linienspezifität der veränderten Zellen beantwortet
werden.
veröffentlicht in:
https://www.future-science.com/doi/10.2144/000113612
nach oben zur Projektseite home
08/5
Identifizierung
von Cofaktoren des Proto-Onkogens Tal 1:
Aufdecken von Zielen für eine molekurare Therapie von Leukämie
Dr. Jörn Lausen,
Chemotherapeutisches Forschungsinstitut Georg-Speyer-Haus, Frankfurt a.
M.
Die Entstehung von Leukämien wird oft durch
genetische Veränderungen zentraler Steuerungsmoleküle der Zelle
verursacht. Zum Beispiel sind häufig Transkriptions-faktoren betroffen,
die für die Regulation der Gen--aktivität in der Zelle wichtig sind.
Der Transkriptionsfaktor Tal1 ist solch ein Schlüsselregulator, er ist
für die Bildung von hämatopoetischen (blutbildenden) Stammzellen absolut
notwendig und ist bei Leukämien durch Mutation fehlreguliert.
Die Fehlexpression von Tal1 ist mit etwa 60 % aller Fälle
kindlicher akuter lymphoider T-Zell Leukämien (T-ALL) assoziiert.
Außerdem steht die Tal1 Expression in Verbin-dung mit dem AML1/ETO
Onkogen mit der akuten myeloischen Leukämie (AML) im Zusammenhang.
Tal1 rekrutiert regulative
Proteinkomplexe, die aus einer großen Anzahl von Faktoren bestehen
können. Die Art dieser beteiligten Cofaktoren bestimmt die Funktion des
Transkriptionsfaktors im zellulären und gene-tischen Kontext. Über die
spezifische Zusammensetzung des Tal1 Komplexes und die beteiligten
Cofakto-ren ist allerdings bisher wenig bekannt. Insbesondere die
beteiligten Faktoren bei Tal1 assoziierten Leukä-mien sind weitgehend
unbekannt.
Für die Entwicklung einer zielgerichteten Therapie von Leukämien ist es
notwendig, die molekularen Mechanismen zu kennen, die für die Funktion
eines fehlregulierten Transkriptionsfaktors wichtig sind.
Ein Beispiel dafür ist die Identifizierung von
Histon-acetylase-Inhibitoren als potentionelle Wirkstoffsubstanz. Die
Kenntnis der beteiligten Cofaktoren von Tal1 könnte für das rationale
Design therapeutischer Substanzen von großem Nutzen sein. Zum Beispiel
könnten Inhibitoren von Interaktionen zwischen Tal1 und identifi-zierter
Cofaktoren entwickelt oder beteiligte enzymatische Funktionen gezielt
beeinfluß werden.
Ziel des Projektes ist es, Cofaktoren von Tal1 durch
Affinitätsaufreinigung und Massen-spektromie zu identifizieren.
Nachfolgend sollen die identifizierten Faktoren funktionell
charakterisiert und auf eine Beteili-gung in der Leukämieentstehung
untersucht werden.
nach oben zur Projektseite home
08/6
Detektion und
Charakterisierung residueller Blasten bei Kindern mit Rezidiv einer
akuten lymphoblastischen Leukämie mittels multiparametrischer Durchflusszytometrie
Prof. Dr. W.-D. Ludwig, Dr.rer. nat.L. Karawajew, Frau Rita Mitlöhner
(Dipl.-Biotech.)
Robert-Rössle-Klinik mit Schwerpunkt Hämatologie, Onkologie u.
Tumorimmunologie,
Universitätsmedizin Berlin Charitè - Campus Buch
Das Ansprechen auf die therapeutische Behandlung der akuten
lymphoblastischen Leukämie (ALL), sowohl bei der Eersterkrankung als
auch bei den Rezidiven, kann bei verschiedenen Patienten einen sehr
uner-schiedlichen Verlauf nehmen. Manche Patienten zeigen ein
verzögertes Therapieansrechen - in diesem Fall spricht man von der sog.
"minimalen Resterkrankung" (MRD=minimal residual disease). Der Nachweis
von MRD steht als ein entscheidender prognostischer Parameter für den
Krankheitsveerlauf im Mittelpunkt vieler altueller klinischer Studien
bei akuten Leukämien.
Bei Patienten nach einem ALL-Rezidiv ist das Ansprechen auf die zweite
Induktionstherapie ein entscheid-dender Parameter. Dabei gewinnt der
MRD-Nachweis mittels multiparametrischer Durchflusszytometrie zunehmend
an Bedeutung und wird als unabhängier Paramaeter zur
Risikostratifizierung für die Behand-lung der ALL-Ersterkrankung
eingesetzt.
Dagegen bleibt das Potential dieser Methode für die Therapieantwort- und
Risikoeinschätzhung bei Kindern mit ALL-Rezidiv weitgehend ungenutzt.
Im Ramen des beantragten Projektes soll der durchflusszytometrische
Nachweis von MRD bei Kindern, die im ALL-REZ 2002 Protokoll behandelt
werden, etabliert werden. Die zusätzlichen Vorzüge der
multipara-metrischen Durchflußzytometrie liegen dabei in der
Möglichkeit, neben der MRD Detektion, die MRD Zellen während der
Therapie umfassend zu charakterisieren.
Diese neue Erkenntnisse können genutzt werden, um mögliche
Resistenzmechanismen bei Kindern mit ALL-Rezidiv aufzudecken und
letzendlich einen Beitrag für die Entwicklung neuer Therapieansätze zur
Leukämiebehandlung zu leisten.
nach oben zur Projektseite home
08/7
Untersuchung von
"Killer-cell-immunoglobulin-like receptor / HLA Mismatchen" zwischen
Spender und Empfängern auf das Überleben nach allogener
Stammzelltransplantation bei Patienten mit akuter myeloischer Leukämie
PD Dr. med. Hans Martin, Leiter der KMT-Einheit, Oberarzt der inneren Medizin, Med. Klinik III, Abt. Hämatologie und Onkologie, Klinikum der J. W. Goethe-Universität Frankfurt am Main
Natürliche Killerzellen (NK-Zellen) sind
Lymphozyten aus dem Knochenmark, die eine sehr wichtige Rolle im
angeborenen Immunsystem spielen und unter bestimmten Umständen in der
Lage sind, Tumorzellen abzutöten. Die zytotoxische Funktion von
NK-Zellen wird durch aktivierende und hemmende Oberflächen-rezeptoren
reguliert. Hierzu gehören wesentlich die inhibierend wirkenden
„Killer-cell-immunoglobulin-like receptors“ (KIR), die an Klasse I
HLA-Antigene auf potentiellen Zielzellen binden. Passen die HLA-Antigene
auf der Zielzelle, wird die zytotoxische Funktion der NK-Zelle gehemmt.
Fehlen passende HLA-Antigene auf potentiellen Zielzellen, können
diese abgetötet werden. Durch diesen KIR/HLA-ligand match oder mismatch
kann die NK-Zelle zwischen eigenen beziehungsweise fremden oder
veränderten Zellen unterscheiden.
Die Bedeutung von NK-Zellen auf das Überleben von Patienten nach allogener Stammzelltransplantation von HLA-misgematchten bzw. haploidenten Spendern konnte von einer Arbeitsgruppe aus Perugia gezeigt werden (Ruggeri L, et al., Science 2002; 295:2097). Patienten mit einem KIR-misgematchten Spender hatten eine signifikant bessere Überlebenschance.
Die Bedeutung von KIR.-Mismatchen bei transplantierten Patienten mit HLA-kompatiblen Spendern ist nach bisherigen Publikationen nicht so eindeutig. Die untersuchten Patientenkohorten waren hinsichtlich Diagnosen und Transplantationsmodalitäten zum Teil sehr heterogen. Möglicherweise spielen in dieser Konstellation auch andere Parameter eine wichtige modulierende Rolle.
In einer ersten eigenen Untersuchung wurde eine homogenere Gruppe von 54 Patienten mit akuter myeloischer Leukämie (AML) nach allogener Stammzelltransplantation eingeschlossen, bei denen Spender-/Patienten-DNA für eine retrospektive KIR-Typisierung unmittelbar zugänglich war. Es zeigte sich ein signifikant besseres Überleben bei den Patienten, die von einem HLA-kompatiblem aber KIR-Ligand misgematchten Spender transplantiert wurden (Richter et al. Bone Marrow Transplant. Supplement EBMT 2008). Dieses Ergebnis erscheint so vielversprechend, dass es unbedingt in weiteren Untersuchungen untermauert werden sollte.
Geplante weitere
Untersuchungen:
Die Analyse des Einflusses von KIR-Mismatchen soll in der eigenen
Patientenkohorte vervoll-ständigt und deren Einfluß näher untersucht
werden.
Als nächste Schritte sind vorgesehen:
1. Erweiterung der Patientenzahl: Im Zeitraum von 1998 bis 2007 wurden 147 AML-Patienten allogen transplantiert. Die KIR-Typisierung in dieser Kohorte soll - ausgehend von den bisher 54 untersuchten Patienten – möglichst vervollständigt werden.
2. Erweiterung der Dokumentation auf weiteren klinischen transplantationsassoziierten Parametern und multivariate Analyse in der gesamten Patientenkohorte.
3. Prospektive Untersuchung des KIR-match bzw. mismatch Status bei zukünftigen allogenen Transplantationen.
4. Vorbereitende Untersuchungen für funktionelle Analysen.
nach oben
zur Projektseite
home
08/8
Erhöhung der Sicherheit zellulärer
Immuntherapien für pädiatrische
und adulte Patienten mit Hochrisiko-Leukämien
Prof. Dr. Boris Fehse, stellvertr. wissenschaftl. Direktor, Dr. Petra Joh-Haus der Frankfurter Stiftung für krebskranke Kinder in Zusammenarbeit mit der Klinik für Pädiatrische Onkologie und Hämatologie der Universität Ffm.
Die allogene Blutstammzeltransplantation (allo-HSCT) stellt für viele
maligne Erkrankungen des Blutsystems (z.B. akute Leukämien, MDS,
Lymphome, Multiples Myelom) die einzige Therapieoption mit einer
lang-fristigen Heilungschance dar.
Allerdings kann es auch nach einer allo-HSCT zu Rezidiven der
Grundkrankheit kommen, die zumeist mit einer äußerst schlechten Prognose
assoziiert sind. Diese Rezidive lassen sich auf die erneute
Proliferation von Leukämiezellen zurückführen, die trotz der
Hochdosis-Therapie im Körper verblieben sind. Besonders bei aggressiven
Hochrisiko-Leukämien kann es sehr schnell nach der allo-HSCT zu einem
hochmalignen Rückfall kommen, der oft eine fatale Prognose hat.
Seit mehr als 15 Jahren ist bekannt, daß die
Infusion von allogenen (Spender-) Lymphozyten das Ptotential besitzt,
residuale maligne Zellen zu erkennen und zu zerstören. Dies führte zur
Etablierung des Konzepts der adoptiven Immuntherapie. Allerdings
erkennen Spenderlymphozyten auch gesundes Empfängergewebe als fremd und
greifen dieses an, was zu einer schweren, potentiell lebensbedrohlichen
Spender-gegen-Wirt Krankheit (graft versus host desease, GvHD)
führen kann.
Dieses Risiko ist besonders hoch, wenn die Blutstammzellen von
halbidentischen (haploidenten) Spendern stammen, die genetischen
Unterschiede also vergleichsweise groß sind. In diesen Fällen kann schon
die Infusion relativ kleiner Mengen von Spender-T-Zellen zu einer
tödlichen GvHD führen.
Um das Entstehen einer lebensbedrohlichen GvHD zu verhindern
wurde Mitte der 90er Jahre ein Konzept vorgeschlagen, das auf der
Ausstattung der Spender-T-Zellen mit einem „Suizidgen“ beruht.
Die Aktivierung des Suizidmechanismus ermöglicht somit im Falle der
Entwicklung einer schweren GvHD die selektive Eliminierung alloreaktiver
T-Lymphozyten.
Aus unterschiedlichen Gründen hat es sich
aufgrund einer Reihe praktischer Schwierigkeiten nicht in großem Maßstab
durchgesetzt. Durch die größere Verbreitung der allogenen
Blutstammzelltransplantation (allo-HSCT) hat das zugrunde liegende
klinische Problem an Schärfe eher zugenommen.
Das oben genannte Projekt hat zum Ziel, das Konzept der mit
Suizidgen-modifizierten T-Zellen so weiter zu entwickeln, daß am Ende
der dreijährigen Förderphase eine klinische Studie bei adulten wie auch
pädiatrischen Hochrisiko-Leukämiepatienten unter wesentlich verbesserten
Voraussetzungen durchgeführt werden kann.
nach oben
zur Projektseite
home
geförderte Projekte 2009
09/1
Analyse der Expression und DNA-Methylierung
von SFRP-1 und SFRP-2 in Knochenmarkzellen von Patienten mit
myelodyspastischem Syndrom (MDS)
und akuter Leukämie (AL)
Prof. Dr. med.
Wolf-K. Hofmann Medizinische Klinik
III Campus Benjamin Franklin Charité - Universitätsmedizin
Hindenburgdamm 30, 2203
Berlin
Das myelodysplastische Syndrom (MDS) ist eine
klonale Erkrankung der Blutbildung, die im Initialstadium durch
ein hyperzelluläres Knochenmark und gleichzeitige periphere Zytopenie
gekennzeichnet ist.
Im weiteren Verlauf geht das MDS oftmals in eine akute myeloische
Leukämie (AML) über. MDS tritt ver-mehrt bei älteren Patienten mit einem
Durchschnittsalter von 65 – 75 Jahren auf. Die Anzahl der
Neuer-krankungen an MDS (Inzidenz) ist in den letzten Jahren
angestiegen.
Die Entstehung des MDS wird auf eine schrittweise Akkumulation genetischer Schäden in hämatopoeti-schen Stammzellen zurückgeführt, die zunächst zu gestörter Differenzierung und gesteigerter Apoptose führt und im weiteren Verlauf durch die Alteration von Zellzyklus-, Proliferations- und Apoptoseassoziierten Genen in die Entwicklung einer AML mündet.
In den letzten Jahren wurden eine Reihe von Tumor-Suppressor-Proteinen, die eine wichtige Bedeutung bei der Kontrolle des Wachstums von normalen Zellen haben, identifiziert.
Darüber hinaus konnte am Beispiel des MDS durch verschiedene zellbiologische und molekulargenetische Methoden gezeigt werden, daß für die leukämische Transformation der hämatopoetischen Stammzelle die Kumulation von einer Vielzahl von endogenen und exogenen Ereignissen erforderlich ist.
Bei den „Secreted
frizzled related proteins“ (SFRPs) handelt es sich um Proteine, die in
einem pathophysio-logisch wichtigen Pathway (WNT-Pathway) zentrale
Kontroll-funktionen übrnehmen.
Epigenetische Veränderungen bei SFRP-1 wurden kürzlich in
AML-Zell-Linien beschrieben, darüber hinaus ist bekannt, daß SFRP-1 eine
differenzierende Funktion bei der normalen Erythropoeseentwicklung
besitzt.
Im vorstehenden Projekt sollen die Expressionen (Ausprägung der genetischen Informationen) und – falls nachweisbar - die DNA-Methylierung1 von S von SFRP-1 und SFRP-2 in hämatopoetischen Stammzellen von MDS und AML/ALL Patienten untersucht werden, um zu klären, ob der Verlust dieser Genfunktion eine Rolle in der Pathogenese des MDS und der akuten Leukämien spielt.
Anmerkung
1
Bei der DNA-Methylierung handelt es sich um eine chemische Abänderung an
Grundbausteinen der
Erbsubstanz einer Zelle. Diese Modifikation wird durch die
Übertragung von Methylgruppen durch
Enzyme an bestimmten Stellen innerhalb der DNA hervorgerufen.
Da der Grundbaustein an der jeweiligen Stelle erhalten
bleibt, ist die DNA-Methylierung keine
genetische
Mutation.
Sie ist eine
DNA-Modifikation,
kommt in sehr vielen verschiedenen (möglicherweise in allen) Lebewesen
vor und hat verschiedene biologische Funktionen.
nach oben zur Projektseite home
09/2
Die Rolle des STAT-.Signalwegs für
die Arsen-vermittelte Apoptose bei AML
PD. Dr./Univ. Perugia Martin Ruthardt, Klinikum der J.W. Goethe-Universität, Med. Klinik III, Hämatologie, Theodor-Stern-Kai 7 60596 Frankfurt,
Bei über 50 % der Patienten mit akuter
myeloischer Leukämie (AML) ist der Transkriptionsfaktor STAT3 aktiviert.
Eine konstitutive Aktivierung von STAT Proteinen ist in der Lage, Zellen
bösartig zu transformieren. Bei der Untergruppe der akuten
Promyelozytenleukämie (AML.M3) wurde in allen bisher untersuchten Fällen
STAT 3 aktiviert gefunden.
Die Besonderheit der AML-M3 ist, dass zwei Substanzen existieren, die
eine Tumorzell-spezifische Behand-lung erlauben, d. h. die gezielt nur
die leukämischen Zellen treffen und nicht Zellen der normalen Gewebe.
Dabei handelt es sich um die all-trans Retinsäure (t-RA), einem
Abkömmling des Vitamin A und um Arsen-trioxid (Arsen) in niedrigen
Dosen.
Die Behandlung mit einer dieser Substanzen als Monothereapie führt in 80
- 90 % der Fälle zu einer hämato-logischen Vollremission. Allerdings ist
nur Arsen in der Lage, eine hohe Rate lang auernder Vollremissionen zu
induzieren, während AML-M3 Patienten, die nur mit t-RA behandelt wurden,
keine molekulare Remission erreichen und unweigerlich innerhalb weniger
Monate rezividieren. Kürzlich wurde gezeigt, dass Arsen über eine
Modifikation der Phosphorylierung in der Lage ist, STAT3 zu hemmen
(Cheng et al., 2004, Wetzler
et al, 2006)
Hier soll nun geklärt werden, welche Rolle die konstitutive Aktivierung
von STAT3 für die Leukämieinduktion spielt und ob die Hemmung STAT3 fr
die Tumorzell-spezifische Wirkung von Arsen bei der AML-M3
verant-wortllich ist und damit auch ein Target bei allen AML Patienten
darstellt, bei denen STAT3 konstitutiv akti-viert ist.
Diese Untersuchungen werden einen Beitrag dazu leisten, die Bedeutung
und die bisher in der Literatur sehr umstrittenen Wirkmechanismen von
Arsen besser zu verstehen. Dies ist von besonderer Bedeutung, da Arsen
möglicherweise eine wichtige Rolle bei der Erhaltungstherapie durch eine
mögliche Unterdrückung der Leukämie-initiierenden Stammzelle zukommen
könnte.
nach oben zur Projektseite home
09/3
Zielgerichtete Erkennung und Zerstörung von
Krebszellen durch natürliche Killerzellen
PD Dr, rer,
nat. Joachim Koch,
Chemotherapeutisches Forschungsinstitut Georg-Speyer-Haus, Ffm.
Das Immunsystem der Wirbeltiere setzt
sich aus der angeborenen und der erworbenen Immunantwort zusammen.
Die Komponenten des angeborenen Immunsystems, wie z. B. toxische Peptide
(Spaltprodukte des Eiweißabbaues), Makrophagen (große
Freßzellen) und natürliche Killerzellen (NK Zellen), reagieren auf
Phathogene (Krankheitsauslöser) innerhalb von Sekunden bis
Minuten, um Infektionen (aber auch Krebs-zellen) einzudämmen bzw. zu
bekämpfen, bevor diese sich manifestieren können oder die erworbene
Immunantwort sich etabliert hat.
NK Zellen vermitteln durch Integration einer Vielzahl von Signalen ihrer
inhibitorischen (hemmenden) oder aktivierenden Rezeptoren
zwischen dem angeborenen und dem erworben-en Immunsystem.
Die Aktivierung von NK Zellen zur Zerstörung von virusinfizierten Zellen
oder Tumorzellen hängt maßgeblich von der Vorherrschaft aktivierender
Signale durch die zentralen humanen aktivierenden NK Zell Rezeptoren
NKp30, NKp44 und NKp46 (NCRs) ab.
Die zellulären Liganden (Atome oder Moleküle, die eine chemische
Bindung bewirken) der NCRs sind bisher unbekannt.
Im Rahmen des vorstehenden Projektes sollen
diese zellulären Liganden daher identifiziert und biochemisch
charakterisiert werden, um einen grundlegenden Vorgang im Immunsystem
aufzuklären.
Darüber hinaus setzen diese Erkenntnisse Impulse für die Etablierung
therapeutisch nutzbarer NK-Zell- Populationen, die Krebszellen
spezifisch erkennen und eliminieren können.
nach oben zur Projektseite home
09/4
Aufbau eines MDS-Zentrums am
Universitätsklinikum Mannheim (UMM)
Prof. Dr. Wolf-Karsten Hofmann, Direktor der III. Med. Klinik Hämatologie und Onkologie, Universitätsmedizin Mannheim, Theodor-Kutzer-Ufer 1 - 3, 68167 Mannheim
Am Universitätsklinikum Mannheim wurde mit der
Etablierung eines
MDS-Zentrums zur Diagnostik und Therapie von Patienten mit
myelodysplastischem Syndrom (MDS)
und
der Errichtung eines wissenschaftlichen Labors zur Untersuchung
der Ursachen dieser Erkrankungen
begonnen.
Da ein solches Zentrum bisher in Mannheim und Umgebung nicht existiert,
kann durch die Einrichtung die Betreuung von Patienten mit MDS bzw.
assoziierten Erkrankungen sicher verbessert werden.
Die Patienten sollen im MDS-Zentrum eine komplette Diagnostik des MDS auf hohem wissenschaftlichen Niveau erhalten. Anschließend werden Krankheits- und Risikospezifische Therapievorschläge erarbeitet und umgesetzt. Die Betreuung der MDS-Patienten erfolgt in unmittelbarer Zusammenarbeit mit den niedergelas-senen Ärzten und den Ärzten in umliegenden Krankenhäusern.
Herr Professor Hofmann
hat von 2004 bis 2009 in Berlin am Universitätsklinikum Benjamin
Franklin ein national und auch international anerkanntes MDS-Zentrum
aufgebaut. Neben der konsequenten Umsetzung der aktuellen Aspekte der
molekularen Diagnostik und krankheitsspezifischen Therapie konnte ein
grund-lagenwissenschaftliches Forschungsprofil auf dem Gebiet des MDS
geschaffen werden.
Mit der Startförderung wurde der Grundstein für die Laborausrüstung
gelegt.
siehe auch Veröffentlichung
Genes, Chromosomes & Cancer 51:756-767 (2012)
nach oben zur Projektseite home
09/5
MLL-Aberrationen bei Leukämien
Dr. med. Dr. rer. nat. Thomas Burmeister Facharzt für Innere Medizin Charité, Campus Benjamin Franklin Med. Klinik für Hämatologie, Onkologie Hindenburgdamm 30, 12200 Berlin
Als „minimale Resterkrankung“ (minimal residual disease, MRD) bezeichnet man den Anteil von Leukämie-zellen nach einer Chemotherapie. Die MRD stellt den wichtigsten prognostischen Faktor bei der akuten lymphatischen Leukämie (ALL) sowohl im Erwachsenenalter als auch im Kindesalter dar. Das Ziel der ALL-Behandlung ist es, die MRD auf ein zuletzt nicht mehr nachweisbares Niveau zu bringen. Patienten, die einen schlechten MRD-Abfall zeigen, haben eine ungünstige Prognose und müssen einer intensivierten Behandlung zugeführt werden.
Patienten mit genetischen Veränderungen des MLL-Gens auf dem langen Arm von Chromosom 11 gelten als „Hochrisiko“-Patienten. Im Allgemeinen wird bei diesen Patienten eine Knochenmark- oder Stammzell-transplantation von einem fremden oder verwandten Spender angestrebt. Bei diesen Patienten sind MRD-Messungen ebenfalls von größter Wichtigkeit sowohl vor als auch nach der Transplantation, da diese Messungen dem behandelnden Arzt wertvolle Hinweise über den Stand der Leukämie geben. Insbesondere kann nach erfolgter Transplantation die notwendige Immunsuppression daran orientiert gesteuert werden.
Im Rahmen des vorliegenden Projektes sollen MRD-Messungen bei erwachsenen MLL-positiven Patienten mit einer neuartigen, vom Antragsteller entwickelten Methode durchgeführt werden. Damit kann eine Leukämiezelle unter 100.000 gesunden Zellen nachgewiesen werden. Damit wird zum einen die Behandlung dieser Patienten unterstützt, zum anderen können wertvolle Informationen über die Genetik des MLL-Gens gewonnen werden.
nach oben zur Projektseite home
09/6
Immuntherapie mit Natürlichen Killerzellen
bei Kindern mit Hochrisiko-Leukämien: Einfluss auf
Leukämie-Immunitätsmechnismen
PD. Dr.Ulrike Köhl, Leiterin des Labors für Stammzelltransplantation und
Immuntherapien Pädiatrische Hämatologie und
Onkologie,Universitätsklinikum der J.W. Goethe-Universität,
Theodor-Stern-Kai 7 60596 Frankfurt,
Natürliche Killer
(NK)-Zellen sind als essentieller Bestandteil des angeborenen
Immunsystems an der Zerstörung von Leukämiezellen beteiligt und eignen
sich daher für eine zelluläre Immuntherapie für Patienten mit
Hochrisiko-Leukämien. Im Rahmen einer laufenden bi-nationalen Studie
(Schweiz/Deutschland) werden Kinder und Erwachsene mit
Hochrisiko-Leukämien nach „haploidenter Stammzelltransplantation“ (Transplantation
mit Stammzellen von den Eltern) zusätzlich mit NK-Zellen ihres
Spenders behandelt. Mit diesem Therapieansatz kann der
Anti-Leukämie-Effekt gesteigert werden. Leider kann dieser Benefit
wieder abgeschwächt werden durch die Freisetzung löslicher Moleküle von
den Leukämiezellen, wodurch die Immunabwehr abgeschwächt wird. Daher
soll im vorliegenden Projekt zunächst die Beteiligung und der negative
Einfluss dieser „Immunitätsmechanismen der Leukämiezellen“ auf die
Zytotoxizität dieser NK-Zellen analysiert werden. Anschließend soll mit
verschiedenen Methoden die NK-Zellfunktion gesteigert werden, um die
Hemmung auf das Immunsystem zu überwinden.
Die
Ergebnisse sollen beitragen, zukünftige Therapien mit NK-Zellen bei der
Bekämpfung von Hochrisiko-Leukämien deutlich zu verbessern und
lang-fristig die Heilungsrate der Patienten zu verbessern.
Forschungsergebnis:
In einer abgeschlossenen Phase I/II Studie mit Natürlichen Killer
(NK)-Zellen zur Behandlung von rezidivierenden Hochrisiko-Leukämien und
Tumoren nach haploidenter Stammzell-transplantation (Deutschland,
Schweiz) konnten wir die Machbarkeit und Verträglichkeit dieser
Zelltherapie zeigen. Die gefährliche „graft versus host disease“ (GvHD =
Transplantat gegen Wirt Reaktion) trat nicht auf, wenn die
kontaminierende T-Zell-Dosis unter 25x10e3 T-Zellen/kg KG lag (Stern M
et al. BMT 2013).
Es zeigten sich aber auch Limitationen in der Therapie durch
„Tumor-Immune-Escape-Mechanismen“. Dabei wurden hohe Plasmaspiegel von
löslichem „class I chain-related peptide A“ (löslMICA) detektiert.
Lösliches MICA bindet an den sogenannten Rezeptor NKG2D auf den
NK-Zellen und kann somit die zytotoxische Aktivität von NK-Zellen
inhibieren. Werden die Spender NK-Zellen aber mit Hilfe des Zytokins
Interleukin 2 (IL-2) aktiviert, fahren sie den Rezeptor NKG2D hoch
(dieser wird dann besonders stark exprimiert). Dadurch sind die
NK-Zellen in der Lage das lösliche MICA abzufangen - mit den noch freien
NKG2D-Rezeptoren können sie die Leukämiezellen erkennen und erfolgreich
zerstören.
Darüber hinaus konnten wir die Herstellung dieser Spender NK-Zellen optimieren für zukünftige Studien.
Eine „GMP“-konforme (GMP=good manufacturing
practica= Reinraumbedingungen) Entfernung der gefährlichen T-Zellen
führte zu einer Reinheit an 94 % CD56+CD3-NK-Zellen.
Die „Clinical
scale“ Expansionsrate bei Stimulation mit 1000 U/ml IL-2 über 10 Tage
lag im Median beim dreifachen. Vergleichende Untersuchungen zur
Stimulierung mit IL-2 versus IL-2 und IL-15 konnten keine Unterschiede
in der Expansionsrate und im intrazellulären Signalling der NK-Zellen
zeigen.
Für Zelltherapeutika, die zur Behandlung von
Patienten eingesetzt werden, ist die Vitalität der kryokonser-vierten
Zellen nach dem Auftauen von besonderer Wichtigkeit.
Wir konnten zeigen, dass unstimulierte NK-Zellen nach dem Auftauen eine
signifikant schlechtere Vitalität zeigen gegenüber kryokonservierten,
IL-2 aktivierten NI-Zellen.
(Koehl U, Frontiers Immunol 2013).
siehe Veröffentlichung:
Front Oncol. 2013 May 17;3:118. doi:
10.3389/fonc.2013.00118. eCollection 2013
Clinical
grade purification and expansion of NK cell products for an optimized
manufacturing protocol.
und
Bone Marrow
Transplant 2013 Mai;48(3):433-8. doi:10.1038/bmt.2012.162.Epub 2012 Sep
3.
und
IL-2 stimulated
but not unstimulated NK cells induce selective disappearance of
pripheral blood cells concomitant restults to a phase I/II study
und
IL-2-activated
haploidentical NK cells restore NKG2D-mediated NK-cell cytotoxicity in
neuroblastoma patients by scavenging of plasma MICA.
nach oben zur Projektseite home
09/7
Untersuchung der globalen DNA-Methylierung
während der In-Vitro Differenzierung von hämatopoetischen Stammzellen
von Patienten mit myelodysplastischem Syndrom
Prof. Dr. Wolf-Karsten Hofmann, Direktor der III. Med. Klinik
Hämatologie und Onkologie, Universitätsmedizin Mannheim,
Theodor-Kutzer-Ufer 1 - 3, 68167 Mannheim
Das gestörte Wachstum von Blutzellen bei Patienten mit
myelodysplastischem Syndrom (MDS) ist offen-sichtlich ursächlich mit
Veränderungen im Bereich der Chromosomen oder aber einzelner Gene
verknüpft. Dies ist wahrscheinlich auch die Ursache dafür, daß es später
zur Entwicklung der bösartigen akuten myeloischen Leukämie kommt.
Eine Vielzahl von Störungen liegt im Bereich der GenInformationen, die in der menschlichen DNA gespei-chert sind, vor. Es gibt verschiedene Veränderungen wie den Verlust von DNA-Abschnitten (Deletionen), Veränderungen des genetischen „Codes“ in den MDS-Zellen (Mutationen), Vervielfachungen von DNA-Abschnitten (zum Beispiel Verdopplungen – Duplikationen) und andere. Außerdem hat man durch systematische Untersuchungen herausgefunden, daß einige Abschnitte der DNA auf eine bestimmte Art und Weise chemisch modifiziert („methyliert“) sind. Im Zusammenhang mit einem Projekt, welches bereits von der Gutermuth-Stiftung gefördert wurde, konnten solche chemisch veränderten Abschnitte auf der DNA von MDS-Zellen gefunden werden.
Das jetzt hier beantragte Projekt setzt diese Arbeiten fort und nutzt eine sogenannte „Chip-Technik“, um die chemische Veränderung (Methylierung) von tausenden Genen in einem Experiment in blutbildenden Zellen beim MDS zu analysieren. Durch den Einsatz dieser neuen Technik könnte es möglich sein, herauszu-finden, ob die DNA und die darin verschlüsselten genetischen Informationen falsch oder gar nicht abgelesen werden, was zu einer dramatischen Störung der Zellteilung und des Wachstums der Blutzellen führen kann. Die Folge ist die beim MDS beobachtete Blutarmut.
Da es inzwischen bereits Medikamente gibt, die die genannte Methylierung der DNA hemmen können, sind die Erkenntnisse aus den hier angestrebten Untersuchungen von hoher direkter Bedeutung für das zukünftige Behandlungskonzept bei Patienten mit MDS.
Weitere Förderung siehe 2010 1
nach oben zur Projektseite home
09/8
Einfluß des Histondeacetylase-Inhibitors Panobinostat auf die mmunrekonstitution nach allogener Stammzelltransplantation
bei Hochrisiko-AML und MDS
PD Dr. Gesine Bug,
Medizinische Klinik II, Abt. Hämatologie/Onkologie Klinikum
der Johann Wolfgang Goethe-UniversitätTheodor-Stern-Kai 7, 60590
Frankfurt a. M.
Durch
Intensivierung der antileukämischen Therapie mit allogener
Stammzelltransplantation konnten die Behandlungsergebnisse auch älterer
Patienten mit akuter myeloischer Leukämie (AML) und myelodysplastischem
Syndrom (MDS) verbessert werden.
Leider bewahrt eine allogene Stammzelltransplantation nur einen Teil der Patienten mit einer Hochrisiko-AML bzw. MDS vor einem Rückfall. Das Rückfallrisiko ist im ersten Jahr nach der allogenen Stammzelltransplantation am größten. Es variiert je nach Art und Stadium der bösartigen Erkrankung und des genauen Transplantationsregimes, kann aber bis zu 40 % betragen. Der Rückfall geht von Leukämiezellen aus, die nach der Chemotherapie und/oder Bestrahlung noch im Körper vorhanden, aber selbst mit sehr empfindlichen Methoden nicht mehr nachweisbar sind. Ein Rückfall entsteht, wenn die im Transplantat enthaltenen Immunzellen des Spenders das Wachstum der Blasten nicht verhindern und somit die bösartige Erkrankung nicht kontrollieren können.
Bei manchen Patienten kommt es andererseits durch überschießende Vermehrung der Immunzellen des Spenders, zu lebensbedrohlichen Entzündungsvorgängen, die man Transplantat gegen Empfänger-Reaktion (GvHD) nennt. Eine GvHD kann bei 15 - 30% der Patienten schwerwiegend oder tödlich sein.
Für den langfristigen Erfolg einer allogenen Stammzelltransplantation ist also entscheidend, sowohl einen Rückfall der AML bzw. des MDS als auch eine schwere GvHD zu verhindern. Eine vielversprechende Strategie stellt der Einsatz von Histondeacetylase-(HDAC-) Inhibitoren nach dosisreduzierter allogener Stammzelltransplantation dar, um die Zeit bis zur Ausbildung einer ausreichenden Blastenkontrolle durch Immunzellen des Spenders zu überbrücken.
Der HDAC-Inhibitor Panobinostat gehört zu einer Klasse neuer, intelligenter Medikamente, die in die Genregulation bösartiger, aber auch normaler Zellen eingreifen. Die Wirkung von Panobinostat beruht auf der Hemmung von Enzymen, den sog. HDAC. HDAC entfernen chemische Gruppen von Kernproteinen und anderen Eiweißmolekülen. Dadurch können bestimmte Gene nicht mehr abgelesen werden, was die Entstehung und das Fortschreiten einer Krebserkrankung begünstigt. Die Hemmung der HDAC durch Panobinostat erneuert die Lesbarkeit dieser Gene und führt zu Wachstumsstillstand und Zelltod der bös-artigen Zellen.
Panobinostat hat sich als effektiv gegenüber Leukämiezellen in Laboruntersuchungen und kleineren Studien mit Patienten erwiesen. Mit Panobinostat nahe verwandte Substanzen wirken im Maus-Transplantations-modell zusätzlich immunregulatorisch, d.h. sie hemmen die GvHD, nicht aber den antileukämischen Effekt.
Wir werden deshalb das Konzept einer Erhaltunsgtherapie mit Panobinostat nach allogener Stammzelltrans-plantation im Rahmen einer klinischen Studie prüfen. Unsere Studie basiert auf der Annahme, daß Panobinostat Patienten mit Hochrisiko-AML oder MDS vor einem Rückfall schützen und gleichzeitig die GvHD-Reaktion bei erhaltenem antileukämischem Effekt minimieren kann. Im Rahmen des von der Alfred und Angelika-Guthermuth-Stiftung geförderten Projekts sollen aufwendige studienbegleitende Untersuchun-gen zur Funktion des Immunsystems nach allogener Stammzelltransplantation durchgeführt werden. .
nach oben zur Projektseite home
09/9
Rolle des dysregulierten Lipocalin-Signalwegs
für Apoptose-Resistenz hämopoetischer Stammzellen unter der Expression
Leukämie-assoziierter Fusionproteine
D. Dr. med. Martin Ruthardt Klinikum der
Goethe-Universität Med. Klinik III, Hämatologie,
Theodor-Stern-Kai 7 60596 Frankfurt a. M.
n der
normalen Hämopoese trägt der programmierte Zelltod (die Apoptose) von
bestimmten Zellen zur korrekten Zusammensetzung des Knochenmarks und
damit zur reibungslosen Blutbildung bei. Bei der Leukämie zeigen die
Leukämiezellen eine gestörte Antwort auf die physiologischen Stimuli der
Apoptoseinduktion und damit eine Resistenz gegen die Apoptose, die zur
Induktion und Erhaltung der Leukämie beiträgt. Wie es zu dieser
Resistenz gegen die Apoptose kommt, ist weitgehend unbekannt.
In Modellen der Leukämie ist ein Weg der Apoptoseinduktion, der durch
den Rezeptor für das Lipocalin 2, dem 24p3R vermittelt wird, durch das
Leukämie-induzierende BCR/ABL Fusionsprotein dereguliert. Dadurch werden
die Zellen, die BCR/ABL tragen, resistent gegen die Apoptose. BCR/ABL
dereguliert die Lipocalin 2 induzierte Apoptose durch Mechanismen, die
auch durch andere Leukämie-assoziierte Fusionsproteine und
Leukämiesubtypen aktiviert sind.
In diesem Projekt soll untersucht werden, ob auch andere
Leukämie-induzierende Fusionsproteine Apoptose auf die gleiche Weise
hemmen wie BCR/ABL und welche Rolle das für die Leukämogenese spielt.
Diese
Untersuchungen und die daraus resultierenden Ergebnisse sind von großer
Bedeutung für ein besseres Verständnis der Mechanismen der Leukämogenese
und damit für die Entwicklung neuartiger Therapieansätze im Kampf gegen
die Leukämie.
http://www.haematologica.com/content/92/4/542.full.pdf
nach oben zur Projektseite home
09/10
Molekulare
Charakterisierung der akuten undifferenzierten und biphänotypischen
akuten Leukämien
PD. Dr. med. Claudia D Baldus, Charité
Universitätsmedizin Berlin, Campus Benjamin Franklin, Hämatologie und
Onkologie, Hindenburgdamm 30,
2203 Berlin
In den letzten Jahren konnten zahlreiche
Forschungsarbeiten zur Aufschlüsselung von molekulargenetischen
Veränderungen wesentlich zum Verständnis der Leukämieentstehung
beitragen. Auf diese Erkennt-nisse aufbauend können nun Patienten mit
akuter Leukämie in verschiedene molekular-definierte Risiko-gruppen
eingeteilt werden. Dies ermöglicht es den Ärzten, die Therapie für den
jeweiligen Patienten ent-sprechend des Risikoprofils der Leukämie
abzustimmen. Darüber hinaus ermöglichen diese neuen
Forschungserkenntnisse die Entwicklung von neuen, spezifischen
Therapiemöglichkeiten.
Die Chemotherapie bildet weiterhin die Basis
einer wirksamen Behandlung. Weitergehende Untersuchungen bei der akuten
myeloischen Leukämie (AML) konnten neue molekulare Marker mit
prognostischer wie therapeutischer Relevanz offen legen. Des Weiteren
zeigen sich ähnliche molekulare Veränderungen bei der akuten
lymphoblastischen Leukämie (ALL).
Somit werden nun zunehmend die chemotherapiebasierten Behandlungen mit
neuen (molekularen) Therapieansätzen ergänzt, die gegen spezifische
Veränderungen der Leukämiezellen gerichtet sind. Hierbei kommen sowohl
für die lymphatischen wie auch myeloischen Leukämien neue Substanzen zum
Einsatz, die aktivierte (mutierte) Signalwege der Leukämiezellen
blockieren. Des Weiteren kommen antikörper-basierte Behandlungen hinzu,
die sich gegen spezifische Oberflächeneigenschaften der Leukämiezellen
richten.
Grundsätzlich lassen sich akute Leukämien entsprechend ihrer Differenzierung in myeloische oder lymphatische Leukämien einteilen. Eine kleine Subgruppe von akuten Leukämien umfasst die akute undif-ferenzierte Leukämie (AUL) und die biphänotypische akute Leukämie (BAL), die nicht eindeutig der myeloischen oder lymphatischen Subgruppe zugeordnet werden können. Dies ist jedoch wesentlich, weil sich die Wahl der Chemotherapie nach dieser Linienzugehörigkeit ausrichtet. Im Gegensatz zu den defi-nierten Subgruppen der AML und ALL, ist die Zuordnung der AUL und BAL und damit die Wahl der Chemo-therapie schwierig. Zudem ist die Prognose der Patienten mit BAL und AUL als ungünstig einzuschätzen, da diese Patienten schlecht auf eine lymphatisch bzw. myeloisch ausgerichtete Therapie ansprechen.
Um eine bessere zielgerichtete Therapieauswahl zur Behandlung der BAL und AUL zu ermöglichen, soll in diesem Forschungsprojekt eine molekulare Charakterisierung der BAL und AUL aufgegriffen werden. Insbe-sondere sollen diese auf Veränderungen untersucht werden, die bereits für die AML und ALL beschrieben sind. Entsprechend könnten so auch für die AUL und BAL zielgerichtete Therapien zum Einsatz kommen. Ziel ist es eine verbesserte Therapie für diese Patienten, die bisher mit einer reinen Chemotherapie nur unzu-reichend behandelt waren, zu erzielen. Diese genaue Charakterisierung von genetischen Veränderungen soll zu einer verbesserten prognostischen wie therapeutischen Eingruppierung der AUL und BAL Subgruppen führen.
nach oben zur Projektseite home
09/11
Durchflusszytometrische Untersuchungen zu
Antikörper-basierten therapeutischen Behandlungen von
Hochrisikopatienten mit Rezidiv einer akuten lymphoblastischen Leukämie
Prof. Dr. W.-D. Ludwig,
Dr. L. Karawajew, Charité Universitätsmedizin Berlin,
Campus Buch, 13125 Berlin
Die Prognose von Kindern mit ALL, die einen Krankheitsrückfall
(Rezidiv) erlitten haben, konnte durch opti-mierte chemotherapeutische
Behandlungsstrategien in der aktuellen ALL-REZ 2002 Studie verbessert
wer-den Dennoch besteht insbesondere für Patienten, bei denen diese
Therapie keinen dauerhaften Erfolg zeigt, dringender Bedarf an neuen
therapeutischen Ansätzen. Sie sollen dazu dienen, den Tumor spezifisch
zu eli-minieren und gleichzeitig die Nebenwirkungen der Behandlung zu
reduzieren.
Für eine solche zielgerichete Therapie eignen sich die sog. Antikörper.
Sie Sind Proteine des Immunsys-tems, die sich spezifisch an Zielmoleküle
auf der Oberfläche der Tumorzellen heften können.
Dieser Vorgang bewirkt, dass das Immunsystem des Patienten die
Krebszellen von anderen unterscheiden und durch variable
Wirkungsmechanismen vernihten kann.
Einige dieser neuen Therapeutika sind bereits zuelassen und al
erfolgreiche Medikament auf dem Markt etablier.
Um die Behandlung mit den Antikörpern zu ermögllichen, sollen
leukämische Zellen vor der Behandlung bezüglich der Expression der
entsprechenden Zielmoleküle untersucht werden. Darüber hinaus sollen die
Veränderungen drs Expression während der Therapie nachverfolgt und
berücksichtigt werden. Mit der sog. multiparametrischen
Durchflusszytometrie (MFC) steht uns für diese Zwecke eine
hocheffiziente analytische Methode zur Verfügung. Mit Hilfe dieser
Methode können mehrere Tausend Zellen innerhalb kürzester Zeit auf die
Expression von bis zu acht Zielmolekülen an der Oberfläche untersucht
werden.
Im Rahmen des beantragten Projektes soll die MFC-Techniuk für die
eingehende Untersuchung der Zell-proben von ALL-Rezidivpatienten vor der
Antikörper-basierten therapeutischen Behandlung eingesetzt werden.
Diese Methode wird es uns auch erlauben, zellbiologische Veränderungen
in der Zelle während und nach der erfolgten Antikörper-Therapie zu
verfolgen. Diese Erkenntnisse werden wesentlich zur Entwicklung und
Optimierung neuer Therapieansätze für die Leukämiebehandlung beitragen.
siehe auch:
http://bloodjournal.hematologylibrary.org/content/115/18/3763.full.pdf+html
nach oben zur Projektseite home