geförderte Projekte 2010
10/1
Untersuchung der globalen DNA-Methylierung während der in-vitro Differenzierung von hämatopoetischen Stammzellen von Patienten mit myelodysplastischem Syndrom
Fortsetzungsförderung des 2009 begonnenen Projektes
Prof. Dr. Wolf-Karsten Hofmann, Direktor der III. Med. Klinik
Hämatologie und Onkologie,
Universitätsmedizin Mannheim, Theodor-Kutzer-Ufer 1 - 3, 68167 Mannheim
siehe auch Projekt (2009 7)
Unmittelbar nach
Etablierung des wissenschaftlichen Labors an der Universitätsmedizin
Mannheim konnten die notwendigen Voraussetzungen geschaffen werden, um
die Methylierungs-Arrays für die Analyse von hämatopoetischen Zellen von
Patienten mit MDS vorzubereiten.
Es wurden erste Proben von gesunden Spendern und von Patienten mit MDS
analysiert.
Die vorläufige Datenanalyse erbrachte erste Hinweise, daß WT1 – ein Schlüsselgen in der hämatopoetischen und besonders myeloischen Differenzierung in MDS-Zellen eine aberrante DNA-Methylierung aufweist.
Aktuell werden die
Ergebnisse der Array-Untersuchung statistisch ausgewertet. Die ersten
Qualitätskontrollen haben gezeigt, daß die Methode robust und
zuverlässig auf klinische Knochenmarkproben angewandt werden kann.
Deshalb besteht eine hohe Motivation, weitere Proben von Patienten mit
MDS zu untersuchen.
siehe:
https://www.future-science.com/doi/10.2144/000113612
10/2
Molekulargenetische Analyse von CD34+ Zellen aus Knochenmark von Patienten mit myelodysplastischem Syndrom mittels "Next Generation Sequencing"
Fortsetzungsförderung des
2006 begonnenen Projektes mit neuer Technik
- Pilotprojekt
-
Prof. Dr. Wolf-Karsten
Hofmann, Direktor der III. Med. Klinik Hämatologie und Onkologie,
Universitätsmedizin Mannheim, Theodor-Kutzer-Ufer 1 - 3, 68167 Mannheim
Es gibt zahlreiche Hinweise dafür, daß die Entstehung eines MDS auf die
Anhäufung genetischer Schäden in blutbildenden Zellen des Knochenmarks
zurückzuführen ist. Über die Art dieser Schäden ist bisher jedoch nur
wenig bekannt.
Kürzlich wurde mit der Einführung von „500K SNP mapping arrays“ eine
sehr robuste Technologie auf der Basis von „DNA-Chips“ entwickelt, die
erstmals eine detaillierte Analyse dieser genetischen Veränderungen
erlaubt. Mit diesen DNA-Chips können kleinste (bisher nicht sichtbare)
Veränderungen am genetischen Material in den Blutzellen dargestellt
werden.
Bisher unklar ist auch, ob die genetischen Veränderungen ausschließlich blutbildende Vorläuferzellen von MDS-Patienten betreffen, oder ob es in den Knochenmarkzellen (die auch ausgereifte Zellen enthalten) gleichfalls signifikante Veränderungen gibt. Weiterhin unklar ist, wie sehr die genetischen Profile der einzelnen Zellarten miteinander im Zusammenhang stehen.
Der Einsatz der SNP-Arrays erfordert allerdings die Kenntnis der einzelnen SNP-Positionen im Genom. Bisher unbekannte Regionen ohne SNP bleiben der Analyse verborgen.
Seit 2009 ist eine neue Technik verfügbar, die im Hochdurchsatzverfahren DNA-Abschnitte sequenzieren, also Base für Base darstellen kann. Die Einführung dieses sog. "Next Generation Sequencing" (NGS) ermöglicht es jetzt, eine Quantifizierung von genetischen Veränderungen festzustellen.
Sie wird als Pilotprojekt zur molekulargenetischen Analyse von CD34+ Zellen von Patienten mit MDS zum Einsatz kommen.
siehe Veröffentlichung: http://jmg.bmj.com/content/50/2/108.short
nach oben
zur Projektseite
home
10/3
Charakterisierung der molekularen und prognostischen Bedeutung von
IGFBP2 bei der akuten myeloischen Leukämie (AML)
Priv. Doz. Dr. med. Claudia Baldus Charité –
Universitätsmedizin Berlin - Campus Benjamin Franklin -Hämatologie und
Onkologie, Hindenburgdamm 30,12203 Berlin
Die Identifizierung von genetischen Veränderungen bei
akuten Leukämien hat in den letzten Jahren wesentlich zur verbesserten
Einschätzung der Überlebenswahrschein-lichkeiten der Patienten
beigetragen. Des Weiteren werden auf dem Boden dieser Erkenntnisse neue
Medikamente entwickelt, die gezielt gegen diese veränderten Signalwege
gerichtet sind.
Diese neuen Substanzen werden nun in klinischen Studien in Kombination
mit einer Standardchemotherapie getestet.
Da der Leukämieentstehung viele verschiedene genetische Veränderungen zu
Grunde liegen, ist es das Ziel der klinischen Forschung, weitere
Risikofaktoren und neue Ansätze für modere Medikamente zu entwickeln.
Bei vielen Tumorerkrankungen konnten Insulin und insulinähnliche Wachstumsfaktoren (IGF System) als wesentlich für die Entstehung und das Fortschreiten von bösartigen Erkrankungen dargestellt werden. Hieraus ergeben sich auch neue Therapieoptionen für Krebserkrankungen, die eine Modulation der IGF-Achse als Ziel haben. So finden diese Substanzen, wie Antikörper oder Tyrosinkinaseinhibitoren, bereits Eingang in die klinische Testung zur Behandlung verschiedener Tumorerkrankungen. Die Bedeutung dieses IGF Systems ist bei akuten Leukämien bisher nur wenig untersucht. Proteine, die in diesem Signalweg regulierend eingreifen sind die Insulin-Growth-Factor-Binding-Proteine (IGFBPs).
Das Ziel dieses Projektes ist es, ein Mitglied dieses IGFs Systems, das IGFBP2, bei Patienten mit akuter myeloischer Leukämie zu untersuchen. Insbesondere soll analysiert werden, ob die IGFBP2 Expression von prognostischer Bedeutung ist, und ob sie mit weiteren klinischen und molekularen Marker assoziiert ist.
High expression of IGFBP2 is associated with chemoresistance in adult acute myeloid leukemia.nach oben zur Projektseite home
10/4
Durchflusszytometrische Untersuchungen zu Antikörper-basierten
therapeutischen Behandlungen von Hochrisikopatienten mit Rezidiv einer
akuten lymphoblastischen Leukämie
Dr.
L. Karawajew, Charité Universitätsmedizin Berlin, Campus Buch, 13125
Berlin
Zwischenfinanzierung einer
Doktorandenstelle. siehe
Projekt 209 11
10/5
Untersuchung der antileukämischen Wirksamkeit des HSP70 Inhibitos Pifithrin-µ bei akuten Leukämien
Priv. Doz. Dr. med. Claudia Baldus Charité – Universitätsmedizin Berlin - Campus Benjamin Franklin -Hämatologie und Onkologie, Hindenburgdamm 30,12203 Berlin
Eine Verbesserung der Behandlung akuter Leukämien erfordert die Charakterisierung von neuen und spezifischen Zielstrukturen auf den Leukämiezellen. Auf dieser Grundlage kann die Entwicklung und die klinische Anwendung von modernen, zielgerichteten Medikamenten weiter vorangetrieben weden.
Einen solch neuen Angriffspunkt für molekular ausgerichtete Therapiekonzepte stellen die Hitzeschock Proteine (Heat shock proteins; HSPs) dar.
HSPs werden bei vielen bösartigen Erkrankungen von den malignen Zellen vermehrt produziert und besitzen eine wichtige Rolle in der Regulation des Zellwachstums. Auch bei akuten Leukämien konnte eine Hochregulation der HSPs in den Leukämiezellen beobachtet und mit einer erhöhten Unempfindlichkeit (Reistenz) gegenüber klassischen Chemotherapeutika assoziiert werden.
Ein Mitglied der HSP Familie, das HSP70, ist von besonderer Bedeutung, da es überwiegend durch Stress-signale aktiviert und recht selektiv in malignen Zellen produziert wird. Entsprechend stelle die Blockierung (Inhibition) von HSP70 einen attraktiven neuen Therapieansatz dar. Zudem konnte erst kürzlich Pifithrin-µ als neuer HSP70Inhibitor charakterisiert werden.
Da bisher keine Untersuchungen zur Wirksamkeit von Pifithrin-µ in Leukämiezellen existieren, ist das Ziel dieses Forschunsprojektes, die Effektivität von Pifithrin-µ an Leukämiezellen wie auch an hämtopoetischen Stammzellen zu untersuchen.
nach oben zur Projektseite home
10/6
Molekulargenetische Analyse von CD34+ Zellen aus Knochemark von Patienten mit myelodysplastischem Syndrom mittels "Next Generation Sequencing"
Fortsetzungsförderung des 2006 begonnenen Projektes mit neuer Technik
Prof. Dr. Wolf-Karsten Hofmann, Direktor der
III. Med. Klinik Hämatologie und Onkologie,
Universitätsmedizin Mannheim, Theodor-Kutzer-Ufer 1 - 3, 68167 Mannheim
Finanzierung einer wissenschaftlichen Doktorandenstelle für ein Jahr
siehe auch Projekt
(2009 7)
Unmittelbar nach
Etablierung des wissenschaftlichen Labors an der Universitäts-medizin
Mannheim konnten die notwendigen Voraussetzungen geschaffen werden, um
die Methylierungs-Arrays für die Analyse von hämatopoetischen Zellen von
Patienten mit MDS vorzubereiten.
Es wurden erste Proben von gesunden Spendern und von Patienten mit MDS
analysiert.
Die vorläufige Datenanalyse erbrachte erste Hinweise, daß WT1 – ein Schlüsselgen in der hämatopoetischen und besonders myeloischen Differenzierung in MDS-Zellen eine aberrante DNA-Methylierung aufweist.
Aktuell werden die
Ergebnisse der Array-Untersuchung statistisch ausgewertet. Die ersten
Qualitätskontrollen haben gezeigt, daß die Methode robust und
zuverlässig auf klinische Knochenmarkproben angewandt werden kann.
Deshalb besteht eine hohe Motivation, weitere Proben von Patienten mit
MDS zu untersuchen.
Veröffentlichung in
BioTechniques, Vol. 50, No. 3,
March 2011, pp. 161–164
https://www.future-science.com/doi/10.2144/000113612
Der Einfluss von Mutationen der Isocitratdehydrogenasen 1 und 2 bei der Entstehung der akuten myeloischen Leukämie
Dr. rer. nat. Michael A.
Rieger, Forschungsinstitut Georg Speyer Haus,
Neueste genomweite Untersuchungen haben bei
über 30% aller Patienten mit akuter myeloischer Leukämie (AML)
Mutationen der Enzyme Isocitratdehydrogenase (IDH) 1 und 2 gefunden.
Völlig ungeklärt ist dabei der Mechanismus, der zur Entstehung der AML
durch diese Enzymmutationen beiträgt, wobei vermutet wird, daß es sich
bei diesen Mutationen um frühe Ereignisse in der Leukämogenese handelt.
Fraglich ist, ob die mutierten Enzyme per se krebserregendes
Potential besitzen, oder ob diese Enzyme als Unterdrücker von Krebs
wirken, die ihre Kontrollfunktion durch diese Mutationen verlieren. Um
neue gezielte Therapieansätze entwickeln zu können, ist es notwendig,
den Einfluß dieser neu identifizierten IDH1/2-Mutationen auf die
Entstehung von Leukämien im Detail zu verstehen.
In diesem Projekt soll die unmittelbare Konsequenz dieser Enzymmutationen auf das zelluläre und molekulare Verhalten von hämatopoetischen Stammzellen auf Einzelzellebene untersucht werden.
Hämatopoetische Stammzellen besitzen nicht nur die außergewöhnliche Fähigkeit, lebenslang in alle verschiedenen Blutzelltypen die differenzieren (Multipotenz), sondern sich fortwährend selbst zu erneuern (Selbsterneuerung), um die Anzahl an Stamm-zellen ein Leben lang aufrecht zu halten.
Die kontinuierliche Beobachtung von
angereicherten Stammzellen und all ihren Nachkommen durch die Anwendung
weltweit einzigartiger Technologien aus Langzeit-Zeitraffermikroskopie
und Einzelzellverfolgung (Tracking) soll Aufschlüsse über die
unmittelbaren zellbiologischen Verhaltensänderungen durch die
unterschiedlichen IDH Mutationen während ihrer Differenzierung auf
Einzelzellebene bringen. Diese Verhaltensänderungen werden wichtige
Hinweise über die zugrunde liegenden molekularen Mechanismen liefern,
welche dann gezielt untersucht werden können.
Die Entstehung von leukämischen Stammzellen durch diese Mutationen und
deren mögliche molekulare Angriffspunkte für eine gezielte Therapie
werden in diesem Projekt sowohl in Zellkulturexperimenten als auch im
Mausmodell erforscht.
Die gewonnenen Erkenntnisse sollen den Weg für neue Therapieansätze für eine effiziente Behandlung von IDH-veränderten Leukämien eröffnen
nach oben zur Projektseite home
Unterstützung des Deutschen Registers für Stammzelltransplantationen (DRST)Das Deutsche Register für Stammzelltransplantationen (DRST), gegründet am 03. 04.1998 in Frankfurt am Main, ist zuständig für die zentrale Erfassung und standardisierte Auswertung aller in deutschen Transplantationszentren nach dem 01.01.1998 durchgeführten Transplantationen und liefert zeitnah wichtige Daten über durchgeführte Transplantationen bei verschiedenen Indikationen.
Damit stehen den Transplantationszentren
wichtige Referenzgrößen zur Beurteilung, wie auch zur Planung weiterer
Aktivitäten und Studien zur Verfügung. Darüber hinaus unterstützt das
DRST die Durchführung von nationalen und internationalen
wissenschaftlichen Studien aktiv.
Es arbeitet eng mit dem europäischen Zentrum (European Group for Blood
and Marrow Transplantation, Maastricht, Niederlande) zusammen. Dort
werden europaweit alle Blutstammzelltransplantationen in einer Datenbank
gespeichert.
Definitionen
Blutstammzellen (BSZ) sind die Ausgangszellen der Blutbildung. Unter Blutstammzelltransplantation (BSZT) versteht man die Übertragung von BSZ von einem Spender auf einen Empfänger mit dem Ziel, daß die übertragenen Zellen im Körper des Empfängers anwachsen und biologische Funktionen ausüben.
Sind Spender und
Empfänger verschiedene Personen, spricht man von allogener BSZT.
Erhält der Patient hingegen seine eigenen BSZ, die er zu einem früheren
Zeitpunkt selbst gespendet hat, zurück, spricht man von autologer
BSZT.
Die bekannteste Form der BSZT ist die "Knochenmarktransplantation".
BSZ können jedoch
auch aus dem Venenblut (nach Vorbehandlung mit einem speziellen
Wachstumsfaktor für die Blutbildung, G-CSF genannt) gewonnen werden und
sogar aus dem Restblut des Mutterkuchens (Plazenta).
Werden diese BSZ-Quellen genutzt, spricht man von Transplantation
peripherer Blutstammzellen bzw. Transplantation von plazentarem
Restblut (Nabelschnurblut).
nach oben zur Projektseite home
geförderte Projekte 2011
11/1
Genomweite Charakterisierung der Early T-Cell
Progenitor Leukämie (ETP-ALL)
PD Dr. med. Claudia Baldus, Charité – Universitätsmedizin Berlin -Campus Benjamin Franklin -Hämatologie und Onkologie-, Hindenburgdamm 30,12203 Berlin
Eine Verbesserung der Behandlung
akuter Leukämien erfordert die genaue Charakterisierung von
spezi-fischen Zielstrukturen der Leukämiezellen, die für die Entwicklung
von neuen zielgerichteten Medikamenten notwendig ist.
Die Weiterentwicklungen der molekulargenetischen Technologien erlauben
neben der Untersuchung von Veränderungen (Mutationen) einzelner Gene
eine umfangreiche Mutationssuche aller Gene in Leukämie-zellen.
Diese neuen genomweiten Analysen können so auch Mutationen in bisher
unbekannten Genen nachweisen.
Diese Technologie soll nun eingesetzt werden, um eine spezielle
Subgruppe der akuten Leukämien, die ETP-ALL als stammnahe Leukämieform,
bezüglich genetischer Veränderungen besser zu charakterisieren.
Die Identifizierung von neuen Genmutationen kann nachfolgend nicht nur
für Patienten mit einer ETP-ALL neue Erkenntnisse erbringen, sondern
möglicherweise auch für Patienten mit stammzellnahen Leukämien anderer
Zelllinien.
Diese molekularen Veränderungen im Sinne von Genmutationen können im
Weiteren Aufschluß über neue Prognosemerkmale geben und neue
Zielstrukturen für spezifische Therapiestrategien darstellen.
Veröffentlichung in Blood. 2013;121(23):4749-52
nach oben
zur Projektseite
home
11/2
Molekulargenetische Analyse von CD34+ Zellen aus Knochenmark von
Patienten mit myelodysplastischem Syndrom mittels „Next Generation
Sequencing“
Prof. Dr.
Wolf-Karsten Hofmann, Direktor der III. Med. Klinik Hämatologie und
Onkologie,
Universitätsmedizin Mannheim, Theodor-Kutzer-Ufer 1 - 3, 68167 Mannheim
- Pilotprojekt zur molekulargenetischen Analyse von CD34+ Zellen -
Es gibt zahlreiche
Hinweise dafür, daß die Entstehung eines MDS auf die Anhäufung
genetischer Schäden in blutbildenden Zellen des Knochenmarks
zurückzuführen ist. Über die Art dieser Schäden ist bisher jedoch nur
wenig bekannt.
Kürzlich wurde mit der Einführung von „500K SNP mapping arrays“ eine
sehr robuste Technologie auf der Basis von „DNA-Chips“ entwickelt, die
erstmals eine detaillierte Analyse dieser genetischen
Veränderungen erlaubt. Mit diesen DNA-Chips können kleinste (bisher
nicht sichtbare) Veränderungen am genetischen Material in den Blutzellen
dargestellt werden.
Bisher unklar ist auch, ob die genetischen
Veränderungen ausschließlich blutbildende Vorläuferzellen von
MDS-Patienten betreffen, oder ob es in den Knochenmarkzellen (die auch
ausgereifte Zellen enthalten) gleichfalls signifikante Veränderungen
gibt. Weiterhin unklar ist, wie sehr die genetischen Profile der
einzelnen Zellarten mit-einander im Zusammenhang stehen.
Der Einsatz der SNP-Arrays erfordert allerdings die Kenntnis der
einzelnen SNP-Positionen im Genom. Bisher unbekannte Regionen ohne
SNP bleiben der Analyse verborgen.
Seit 2009 ist eine neue Technik verfügbar, die im Hochdurchsatzverfahren DNA-Abschnitte sequenzieren, also Base für Base darstellen kann. Die Einführung dieses sog. "Next Generation Sequencing" (NGS) ermöglicht es jetzt, eine Quantifizierung von genetischen Veränderungen festzustellen. Sie wird als Pilotprojekt zur molekular-genetischen Analyse von CD34+ Zellen von Patienten mit MDS zum Einsatz kommen.
Veröffentlichung:
Genes, Chromosomes
and Cancer
Volume 51, Issue 8,
pages 756–767,
August 2012
nach oben
zur Projektseite
home
11/3
Untersuchung der globalen DNA-Methylierung
bei Patienten mit akuter Promyelozytenleukämie (APL)
Dr. med. Mark
Reinwald, III. Medizinische Klinik Hämatologie und Onkologie
Universitätsmedizin Mannheim, Theodor-Kutzer-Ufer 1 – 3, 68167 Mannheim
Die akute Promyelozytenleukämie (APL) ist
eine Unterform der akuten myeloischen Leukämien (AML) und macht einen
Anteil von ca. 10-15 % an diesen aus.
Unbehandelt führt die Erkrankung typischerweise innerhalb von wenigen
Wochen zum Tod, charakteristisch sind hierbei schwere Einblutungen mit
nur schwer kontrollier-baren Gerinnungsstörungen.
Ein klassisches Merkmal der APL ist eine genetische Veränderung die zur
Bildung eines Eiweißes namens PML-RARα führt, welches maßgeblich an der
Entstehung und des klinischen Verlaufs der Erkrankung beteiligt ist.
Dieses Eiweiß kann heutzutage mit einer Substanz namens
All-Trans-Retinol-Säure, in Kombination mit einer Chemotherapie gezielt
behandelt werden. Dies stellt in der Leukämiebehandlung einen
einzigartigen Umstand dar, da hierbei die Zellen differenziert, d.h. aus
einem unreifen in den reifen Zustand überführt werden können, und dann
im Verlauf absterben.
Trotz dieser guten Behandlungsmöglichkeit versterben trotzdem an der
Erkrankung sowie Komplikationen ein nicht unerheblicher Teil der
Patienten bzw. erleiden ein Rezidiv, einen Rückfall der Erkrankung. Es
sind bisher nur wenige Faktoren bekannt, die die Wahrscheinlichkeit
eines Ansprechens bzw. eines Rückfalls sowie die insgesamte Prognose von
Patienten mit APL beeinflussen, insbesondere ist nahezu nichts über die
sogenannte DNA-Methylierung der Leukämiezellen bekannt.
DNA-Methylierung ist ein Prozess bei dem die genetische Information von
Zellen modifiziert wird und ent-scheidend an der Steuerung von
Zellwachstum, programmiertem Zelltod, Reifung sowie Zellentwicklung
beteiligt ist.
Es ist bekannt, dass Gene, die Zellwachstum unterdrücken sollen, in
Tumorzellen stark methyliert sind was zur „Abschaltung“ dieser
„natürlichen Wachstumshemmung“ führt. Diese vermehrte DNA-Methylierung
führt somit zu einer Begünstigung zellwachstums-fördernder Prozesse und
hat somit entscheidenden Einfluss an der Tumorentstehung bzw.
-Progression. Über das Ausmaß der DNA-Methylierung bzw. welche Zielgene
bei der APL verstärkt methyliert sind, ist bisher nahezu nichts bekannt.
Das hier beschriebene Projekt soll nun anhand einer neuartigen
array-basierenden Hochdurchsatz-Analyse das Methylierungsmuster bzw.
Ausmaß der globalen DNA-Methylierung in APL-Patienten im Vergleich zu
gesunden Stammzellen untersuchen.
Die gewonnenen Erkenntnisse sollen helfen, einerseits Fortschritte in
der prognostischen Einschätzung als vielleicht auch in Zukunft weitere
therapeutische Möglichkeiten für Patienten mit akuter
Promyelozyten-leukämie zu eröffnen.
11/4
Einfluß des immunsuppressiven Medikaments Mycophenolat-Mofetil auf die Funktionalität von NK-Zellen
PD Dr. Ulrike Köhl, Klinikum der Johann Wolfgang
Goethe-Universität Ffm., Zentrum für Kinder- und Jugendmedizin Klinik
II//III, Leitung des Labors für Stammzelltransplantation und
Immuntherapien,
Theodor Stern Kai 7, 60596 Frankfurt
Die Prognose für einige pädiatrische Patienten mit Hochrisiko-Leukämien ist auch nach allogener Stammzelltransplantation (SZT) schlecht. Daher versuchen wir derzeit in einer klinischen Phase I/II Studie mit allogenen Natürlichen Killer (NK)-Zellen nach haploidenter SZT (Kinder erhalten Stammzellen und NK-Zellen von Vater oder Mutter) den sogenannten Graft-versus-Leukämie (GvL) Effekt (NK-Zellen zerstören
Leukämiezellen) zu verstärken.
NK-Zellen zeigen eine hohe Zytotoxizität gegenüber verschiedenen Leukämien. Nach Stammzelltrans-plantation (SZT) müssen die Patienten über einen längeren Zeitraum immunsuppressive Therapie erhalten, um eine Abstoßung des Transplantats zu verhindern. Dies kann leider auch zu einer verminderten Funktion der NK-Zellen gegenüber Infektionen und gegenüber den Leukämiezellen führen.
Daher ist es Ziel des Projektes, die Wirkung des immunsuppressiven Medikaments Mycophenolat-Mofetil (MMF) auf NK-Zellen zu untersuchen. Des Weiteren soll geklärt werden, ob eine mögliche Hemmung der zellulären Immunfunktion durch MMF reversibel ist. Neben Untersuchungen zur Zytotoxizität der NK-
Zellen sollen auch die intrazellulären Signalwege der NK-Zelle bei MMF-Gabe untersucht werden.
Letztendlich soll versucht werden, für zukünftige Immuntherapien mit NK-Zellen das Behandlungskonzept zu optimieren.
nach oben zur Projektseite home
11/5
Untersuchung von Genpolymorphismen, die möglicherweise einen Einfluss auf das Risiko für die Entwicklung einer akuten lymphatischen Leukämie beinhalten
PD Dr. med. Dr. rer. nat. Thomas
Burmeister, Facharzt für Innere Medizin/Hämatologie,
Onkologie Charité CBF, Medizinische Klinik für Hämatologie/Onkologie,
12200 Berlin Hindenburgdamm 30,
Die akute lymphatische Leukämie (ALL) ist eine vergleichsweise seltene
Erkrankung. Jedoch zeigt sie bezüglich der Häufigkeit eine
charakteristische Altersverteilung und ist im Kindesalter die häufigste
maligne Erkrankung überhaupt.
Die Ursachen für das Auftreten einer ALL und auch die charakteristische
Altersvertei-lung sind letztlich nicht gut verstanden. Eine Vielzahl von
genetischen Veränderungen wurden bei ALL beschrieben, jedoch ist unklar,
warum die Veränderungen bei be-stimmten Individuen auftreten und bei
anderen nicht.
Bezüglich der Entstehung der ALL im Kindesalter wurde die Hypothese
formuliert, dass Infektionen im frühen Kindesalter protektiv
hinsichtlich der späteren Entwicklung einer ALL sein könnten. Diese
Hypothese wird durch einige epidemiologische Studien ge-stützt, ist aber
nicht unumstritten. Eine mögliche Erklärung für den Einfluß der "Umwelt“
auf das Entstehen von Leukämien ist die unterschiedliche genetische
Ausstattung ver-schiedener Individuen. Manche Gene des Immunsystems oder
des Metabolismus von
Fremdsubstanzen weisen Variationen („Polymorphismen“) auf, die
möglicherweise dazu führen könnten, dass die Träger dieser
Polymorphismen unterschiedlich gut auf Umwelt-„Stress“ (Infektionen,
Schadstoffe, .) reagieren können. Dadurch könnten unter Umständen
pathologische Prozesse angestoßen werden, die in eine maligne
Erkrankung, z. B. einer Leukämie münden.
Im Rahmen dieses Projektes sollen bei erwachsenen Patienten mit ALL zwei
Gene auf solche Polymor-phismen untersucht werden: IKZF1 und
ARID5B. Ziel ist es, möglicher-weise Risikofaktoren für das
Auftreten einer ALL im Erwachsenenalter zu identifizieren bzw. generell
dem Verständnis der Pathogenese dieser Erkrankung näher zu kommen
nach oben
zur Projektseite
home
11/6
Unterstützung des Deutschen Registers für Stammzelltransplantationen (DRST)
Das Deutsche Register für Stammzelltransplantationen (DRST),
gegründet am
03. 04.1998 in Frankfurt am Main, ist zuständig für die zentrale
Erfassung und standardisierte Auswertung aller in deutschen
Transplantationszentren nach dem 01.01.1998 durchgeführten
Transplantationen und liefert zeitnah wichtige Daten über durchgeführte
Transplantationen bei verschiedenen Indikationen.
Damit stehen den Transplantationszentren
wichtige Referenzgrößen zur Beurteilung, wie auch zur Planung weiterer
Aktivitäten und Studien zur Verfügung. Darüber hinaus unterstützt das
DRST die Durchführung von nationalen und internationalen
wissenschaftlichen Studien aktiv.
Es arbeitet eng mit dem europäischen Zentrum (European Group for Blood
and Marrow Transplantation, Maastricht, Niederlande) zusammen. Dort
werden europaweit alle Blutstammzelltransplantationen in einer Datenbank
gespeichert.
nach oben
zur Projektseite
home
11/7
Mutatonsanalyse von U2AF35 in Knochenmarkzellen von Patienten mit
myelodysplastischem Syndrom
Prof.
Dr. Wolf-Karsten Hofmann, Direktor der III. Med. Klinik Hämatologie und
Onkologie,
Universitätsmedizin Mannheim, Theodor-Kutzer-Ufer 1 - 3, 68167 Mannheim
Das Myelodysplastische Syndrom (MDS) ist eine klonale Erkrankung der
Blutbildung, die im Initialstadium durch ein hyperzelluläres Knochenmark
und gleichzeitige periphere Zytopenie gekennzeichnet ist. Im weiteren
Verlauf geht das MDS oftmals in eine Akute Myeloische Leukämie (AML)
über.
Die Entstehung eines MDS ist ursächlich mit der
Akkumulation genetischer Schäden in hämatopoetischen Zellen des
Knochenmarks verbunden. Eine zentrale Rolle bei der Steuerung des
Zellwachstums spielt das sogenannte RNA-Splicing, die Modifikation der
genetischen Informationen beim Umschreiben der DNA (Trägerin
der Erbinformation für die Herstellung der RNA)
in RNA
.
Als Spleißen bzw. Splicing (v. engl. splice "miteinander verbinden",
"zusammenkleben") wird ein wichtiger Schritt der Weiterverarbeitung
(Prozessierung) der Ribonukleinsäure (RNA) bezeichnet, Wesentliche
Funktion der RNA in der biologischen Zelle ist die Umsetzung von
genetischer Information in Proteine.
Eine Störung dieses Vorgangs kann zu fehlerhaften RNA-Molekülen und damit zu funktionslosen Proteinen führen. Die Folge davon kann ungesteuertes Wachstum und Vermehrung (Proliferation) von klonalen hämatopoetischen Zellen sein.
In enger Kooperation mit
einer japanischen Arbeitsgruppe bei der Analyse von Genen, die in den
Signalweg des RNA-Splicings eingebunden sind, wurden kürzlich
verschiedene Mutationen entdeckt.
Das vorliegende Projekt wird die seit Juni 2010 in der Klinik etablierte
neue Sequenzierungstechnik „Next Generation Sequencing“ nutzen, die eine
quantitative Mutations-Analyse von U2AF35 in
Knochen-markzellen von MDS-Patienten und anschließend ein Screening für
diese Mutationen ermöglicht.
Seit Etablierung der Technik des „Next Generation Sequencing“ wurde
intensiv an der Etablierung und Optimierung dieses Verfahrens gearbeitet
(federführend Herr Dr. med. Daniel
Nowak).
Durch die Analyse des U2AF35-Gens soll Aufschluss geschaffen werden, ob und wie Veränderungen an diesem Gen zur Patophysiologie und Entwicklung eines MDS beitragen können.
nach oben zur Projektseite home
11/8
Dokumentatonsprojekt zur Praxis der Therapie der akuten Graft versus
Host Disease an deutschen Transplantatonszentren
Dr. Daniela Heidereich / PD Dr.
Stefan A. Klein, Universitätsmedizin Mannheim,
III. Med. Klinik Theodor-Kutzer-Ufer 1 -3, 68167 Mannheim
Die akute Graft versus Host Disease (aGvHD) ist die häufigste
schwerwiegende Komplikation nach allogener Stammzelltransplantation. Es
handelt sich dabei um eine von weißen Blutkörperchen des Spenders, den
sog. T-Lymphozyten vermittelte Reaktion gegen gesundes Gewebe des
Empfängers. Von dieser Reaktion ist häufig der Darm betroffen. Die
Patienten leiden unter schwersten Durchfällen und Bauch-schmerzen.
Besonders bei Patienten mit Beteiligung des Darmes ist der Verlauf der
aGvHD ungünstig und die Sterblichkeit hoch.
Für die Therapie und das Gesamtmanagement der aGvHD existiert kein einheitliches Vorgehen. Zwischen den einzelnen Transplantationszentren unterscheiden sich die Vorgehensweisen erheblich, teils existiert selbst innerhalb eines Zentrums kein Konsens zur Therapie der akuten GvHD. Diese unbefriedigende Situation ist nicht zuletzt Folge des Umstandes, dass es keine fundierten wissenschaftlichen Studien-ergebnisse zur aGvHD gibt. Bestimmend für die heutige Vorgehensweise sind Studien, die vor mehr als 20 Jahren durchgeführt wurden. Allogene Stammzelltransplantationen in den achtziger und frühen neunziger Jahren unterscheiden sich allerdings beträchtlich von der heutigen Praxis. Das damalige Management der akuten GvHD kann somit nicht auf heute übertragen werden.
Um die Grundlage für ein einheitliches Vorgehen sowie für prospektive Studien zur Therapie der aGvHD zu legen, ist es das Ziel, mittels einer retrospektiven multizen-trischen Analyse den tatsächlichen Status quo des Managements und der Therapie zu erfassen. Darüber hinaus sollen an Hand eines großen Patienten-kollektives Risikofaktoren und optimale Therapiestrategien identifiziert werden.
Im Rahmen einer retrospektiven Analyse der Salvagetherapie der Steroid-refraktären aGvHD des Darmes mittels Pentostatin wurden bereits die notwendigen Strukturen etabliert und Daten mittels einheitlicher Dokumentation an acht teilnehmenden Transplantationszentren durch eine Dokumentationsassistentin aus Mannheim erhoben.
In Kooperation mit dem Deutschen Register Stammzelltransplantation (DRST) sollen an Hand der Datensätze aus den teilnehmenden Zentren alle Patienten mit aGvHD im Zeitraum von 2006 bis 2009 mit AML als Grunderkrankung identifiziert werden. Die aGvHD-spezifischen Daten werden anschließend nach einheitlichen Kriterien von einer Person an allen Zentren vor Ort dokumentiert. Es sollen ca. 500 Patienten eingeschlos-sen werden. Die Dokumentationsphase soll sich über das ganze Jahr 2012 erstrecken.
siehe auch Veröffentllichung in
British Journal of Haematology
Volume 154,
Issue 1, pages
143–146, July 2011
nach oben zur Projektseite home
11/9
Detektion und klinische Bedeutung nicht-leukämischer hämatopetischer Vor-läuferzellen bei der Behandlung von Kindern mit Rezidiv einer akuten lympho-blastischen Leukämie
Dr.Leonid Karawajew, Dr. Peter Rhein, Universitätsmedizin Berlin, Klinik für Pädiatrie m. S. Onkologie/Hämatologie, Charité Campus Virchow-Klinikum, Augustenburger Platz 1, 13353 Berlin
Mitfinanzierung einer Stelle eines wissenschaftlichen Mitarbeiters
In der Studie zur Behandlung von Kindern und Jugendlichen mit ALL-Rezidiv intReALL 2010 soll die multiparametrische Durchflußzytometrie als Methode zum Nachweis der sogenannten „minimalen Resterkrankung“ (MRD = minimal residual disease) eingesetzt werden.
Die Identifizierung leukämischer Blasten basiert
auf der Messung mehrerer Proteine (Antigene) auf der Zelloberfläche oder
in der Zelle mittels Antigen-spezifischer Antikörper.
Ein wesentliches Merkmal der multiparametrischen Durchflußzytometrie
besteht darin, dass nicht nur residuale Leukämiezellen sondern auch
andere Zellsubpopulationen, wie reife Leukozyten und normale unreife
hämatopoetische Vorläuferzellen, in den Knochenmark- oder Blut-Proben
nachgewiesen werden.
Die hämatopoetische
Regeneration setzt normalerweise einige Tage nach dem Beenden eines
Therapie-blocks im ALL-REZ BFM Protokoll ein.
Die aktuell bei der multiparametrischen Durchflußzytometrie eingesetzten
8- und 9- Antikörper-Paneels erlauben nicht nur die Identifizierung von
Vorläuferzellen, sondern auch eine detaillierte Beschreibung des
Regenerationsstatus. So können sehr frühe, frühe und späte
Reifungsstufen identifiziert werden.
Die klinische Bedeutung des Regenerationsstatus der der ALL-Rez
Patienten ist jedoch nicht bekannt und wurde bisher nicht systematisch
untersucht.
Unsere bisherige Analyse der MRD-Proben einzelner Patienten zeigt eine
sehr starke, individuelle Variabilität hinsichtlich der Intensität und
Reifungsstufe der hämatopoetischen Regeneration.
Im beantragten Projekt sollen bei den MRD-Untersuchungen die
nicht-leukämischen Vorläufersub-populationen identifiziert und
quantifiziert werden.
Der in dieser Form erfasste Regenerationsstatus
der Patienten wird mit klinischen Daten wie Therapie-antwort und
klinischem Verlauf verglichen um z. B. den Effekt von bestimmten
Medikamenten auf die Regeneration zu untersuchen aber auch auf einen
möglichen Zusammenhang mit der Therapie assoziierten Toxizität zu
überprüfen.
Diese Erkenntnisse sollen wesentlich zur Optimierung bzw. Entwicklung
individualisierter Therapieansätze für die Leukämiebehandlung
beitragen.
nach oben
zur Projektseite
home
11/10
Unterstützung der weiteren Ausbildung
einer mediznisch-technischen Assistentin
mit einem
Stipendium
Einer im Praktikum befindlichen
medizinisch-technischen Assistentin wurde die weitere Ausbildung in
England ermöglicht.
Nach erfolgreicher Weiterbildung ist sie seit 01. 01. 2012 im Labor der
III. Med. Klinik, Hämatologie und Onkologie der Universitätsmedizin
Mannheim angestellt.
nach oben
zur Projektseite
home
12/1
Bedeutung der Cadherine Fat1 und FAT3 bei akuten Leukämien
Prof. Dr. med. Claudia Baldus Charité –
Universitätsmedizin Berlin - Campus
Benjamin Franklin - Hämatologie und Onkologie, Hindenburgdamm 30, 12203
Berlin
Die Verbesserung in der Behandlung von
akuten Leukämien (Blutkrebs) erfordert die Identifizierung von neuen
Molekülen, die als Ansatzpunkte für neue Therapien und als
Risikofaktoren (zur Vorhersage des Rückfallrisikos) bei akuten Leukämien
herangezogen werden können. Insbesondere durch die Weiter-entwicklung
von neuen Untersuchungsmethoden und Technologien ist mittlerweile eine
genomweite Untersuchung von genetischen Veränderungen möglich geworden.
So konnten in den letzten Jahren verschiedenen
Genveränderungen bei akuten Leukämien nachgewiesen werden, wobei die
Bedeutung für eine Vielzahl dieser Veränderungen noch unbekannt ist. In
Vorarbeiten konnten wir Genveränderungen (Mutationen) bei Patienten mit
einer T-lymphoblastischen Leukämie in den Genen FAT1 und FAT3
nachweisen. Diese Gene gehören zu zellulären Oberflächenproteinen
(Cadherine), die eine Vernetzung zwischen verschiedenen Zellen
ermöglichen.
In weiterführenden Untersuchungen soll nun die Expression der Gene FAT1
und FAT3 in Leukämiezellen von Patienten mit akuter Leukämie untersucht
werden. Zudem soll untersucht werden, inwieweit die Cadherine FAT1 und
FAT3 Einfluss auf die Interaktion zwischen Leukämiezellen und umgebenden
Knochenmarkbindegewebe nehmen und durch z. B. eine verstärkte Vernetzung
der Leukämiezellen mit dem umgebenden Gewebe einen ungewollten
Schutzmechanismus für die Leukämiezellen gegenüber der Chemotherapie
darstellen.
veröffentlicht in Blood Cancer Journal (2014) 4, e224; doi:10.1038/bcj.2014.44
nach oben
zur Projektseite
home
12/2
Veränderung in der globalen DNA-Methylierung
bei Patienten
mit MDS und Deletion am Chromosom 5q
Prof. Dr.
Wolf-Karsten Hofmann, Direktor der III. Med. Klinik Hämatologie und
Onkologie,
Universitätsmedizin Mannheim, Theodor-Kutzer-Ufer 1 - 3, 68167 Mannheim
Bei Patienten mit myelodysplastischem Syndrom
werden in ca. 50% der Fälle chromosomale Verände-rungen, d.h.
Veränderungen im Erbgut der blutbildenden Zellen, gefunden. Besonders
häufig ist ein Defekt am 5. Chromosom (Chromosom 5q), welcher typisch
für eine schwere Störung der roten Blutbildung bei Patienten mit MDS
ist. Die ausgeprägte Blutarmut bei diesen Patienten konnte bisher
ausschließlich mit Transfusionen behandelt werden, seit einigen Jahren
steht jedoch ein sehr gut wirksames Medikament (Lenalidomid) zur
Verfügung, mit welchem diese Patienten erfolgreich therapiert werden
können. Dabei gelingt es, bei mehr als 70% eine deutliche Verbesserung
der Blutbildung zu erreichen.
Der Wirkmechanismus dieses neuartigen Medikamentes auf die blutbildenden
Zellen des Knochenmarkes ist bisher nicht genau verstanden. Verschiedene
Untersuchungen haben sich damit beschäftigt, ob die Veränderung am
Chromosom 5 direkt die Störung der Blutbildung verursacht. Bisherige
Ergebnisse zeigen, dass das wahrscheinlich nicht der alleinige Grund
ist. Vielmehr geht man aktuell davon aus, dass durch den Defekt am
Chromosom 5 weitere (sogenannte sekundäre) molekulargenetische
Veränderungen, zum Bei-spiel Störungen der DNA-Methylierung, induziert
werden.
Im vorliegenden Projekt werden deshalb bestimmte Abschnitte der DNA bei
Patienten, die an einem MDS leiden und einen Defekt am Chromosom 5
haben, mittels hochauflösender Methylierungschips analysiert. Die dabei
gefundenen Veränderungen könnten Hinweise auf die Ursache der Störung
der Blutbildung und auf den Mechanismus der Wirksamkeit von Lenalidomid
bei Patienten mit Defekt am Chromosom 5 geben.
nach oben
zur Projektseite
home
12/3
Untersuchung der globalen DNA-Methylierung in
verschiedenen Differenz-ierungsstufen der Hämatopoese zur
Charakterisierung des Methylierungsprofils gesunder Zellen zum Vergleich
mit dysplastischen und malignen Zellen der Hämatopoese
Dr. med. Mark Reinwald, III. Medizinische Klinik
Hämatologie und Onkologie
Universitätsmedizin Mannheim, Theodor-Kutzer-Ufer 1 – 3, 68167 Mannheim
DNA-Methylierung ist ein Prozess bei dem die genetische Information von
Zellen modifiziert wird und somit entscheidend an der Steuerung von
Zellwachstum, programmiertem Zelltod, Reifung sowie Zellentwicklung
beteiligt ist.
Es ist bekannt, das sich während des
Heranwachsens des Embryos (Embryogenese) in Säugetieren eine deutliche
Veränderung des Methylierungsmusters im Rahmen dieses Reifungs- oder
auch Differenzierungs-prozesses zeigt.
Im Mausmodell konnte klar demonstriert werden, dass es insbesondere in
hämatopoetischen Zellen während der Differenzierung zu einer deutlichen
Hypomethylierung bestimmter Genbereiche kommt und dass bestimmte
Reifungsstufen charakteristische Methylierungsprofile aufweisen. Auch
zeigte sich, dass hämatopoetische Stammzellen ein charakteristisches
Methylierungsprogramm aufweisen, welches die Ausdifferenzierung
behindert und die Pluripotenz erhält.
Der Grossteil dieser Studien, der das Methylierungsmuster während der Ausreifung der verschiedenen Zellen der Hämatopoese untersucht, sind aber nur tierexperimentelle Studien, an humanen Zellen wurden solche Experimente bisher nur wenig untersucht.
In dem hier vorliegenden Projekt sollen nun
anhand von menschlichen Zellen, gewonnen aus Stammzell-präparaten, das
Methylierungsmuster während verschiedener Reifungsstufen der
Hämatopoese, beginnend bei der pluripotenten Stammzelle bis hin zur
fertigen, ausgereiften weissen Blutzelle untersucht und näher
charakterisiert werden.
Die somit gewonnenen Erkenntnisse sollen helfen, den Einfluss der
DNA-Methylierung auf den Reifungs-prozess während der physiologischen
Ausreifung von Blutzellen näher zu bestimmen.
Dieses gesunde bzw. physiologische Methylierungsprofil hämatopoetischer Zellen kann dann dazu genutzt werden, bösartige hämatopoetische Erkrankungen wie akute Leukämien oder auch das myelodysplastische Syndrom anhand einer Veränderung im Methylierungsprofil klar abzugrenzen und somit auch Erkenntnisse über die Pathogenese dieser Erkrankungen zu erhalten.
nach oben
zur Projektseite
home
12/4
Untersuchung des
Methylisierungsmusters von überexprimierten Tyrosinkinasen bei der
akuten lymphatischen Leukämie des Erwachsenenalters
PD Dr. med. Dr. rer. nat. Thomas Burmeister,
Facharzt für Innere Medizin/Hämatologie, Onkologie
Charité CBF, Medizinische Klinik für Hämatologie/Onkologie, 12200
Berlin Hindenburgdamm 30
Anschlussfinanzierung der Stelle einer MTA
Bei vielen Tumorerkrankungen spielen sogenannte
Tyrosinkinasen eine Schlüsselrolle. Tyrosinkinasen sind Enzyme,
die an die Aminosäure Tyrosin von anderen zellulären Proteinen ein
Phosphat „anhängen“ und damit bestimmte Stoffwechselwege oder
Signalkaskaden in der Zelle in Gang setzen können. Diese Erkenntnis aus
der Grundlagenforschung ist bereits auch vielfach in den klinischen
Alltag der Behandlung von Tumorpatienten eingegangen
und hat zur Entwicklung von Medikamenten geführt, mit denen
Tyrosin-kinasen selektiv gehemmt werden können.
Diese sogenannten Tyrosinkinase- Inhibitoren (TKI) haben die
Behandlung einiger Tumorerkrankungen in den letzten Jahren geradezu
revolutioniert.
Das Expressionsmuster von Tyrosinkinasen, d. h
letztlich die Konzentration dieser Enzyme in der Zelle, kann durch
verschiedene Prozesse beeinflusst sein, zum einen durch genetische
und zum anderen durch epigenetische Veränderungen. Zu den
letztgenannten zählen Methylierungen von Steuersequenzen („Promotoren“)
der Gene.
Durch den prinzipiell reversiblen Prozess der Methylierung kann die
Expression eines Gens beeinflusst werden. Ein Gen, dessen Promotoren
stark methyliert sind, wird gar nicht oder kaum exprimiert, d. h. kaum
„abgelesen“ und das zugehörige Protein liegt nur in geringer
Konzentration in der Zelle vor. Ein Gen, dessen Promotoren kaum
methyliert sind, wird dagegen in der Regel stark exprimiert, d. h. es
liegt in hoher Konzentration vor.
Im Rahmen dieses Projektes soll der Einfluss des Methylierungsmusters auf die Expression von Tyrosin-kinasen bei Patienten mit akuter lymphatischer Leukämie (ALL) untersucht werden. Das Projekt ist als Anschlussprojekt eines durch die DFG geförderten Projektes gedacht.
nach oben
zur Projektseite
home
12/5
Unterstützung des Deutschen Registers
für Stammzelltransplantationen DRST
Das Deutsche Register für
Stammzelltransplantationen (DRST), gegründet am 03. 04.1998 in Frankfurt
am Main, ist zuständig für die zentrale Erfassung und standardisierte
Aus-wertung aller in deutschen Trans-plantationszentren nach
dem 01.01.1998 durchgeführten Transplantationen und liefert zeitnah
wichtige Daten über durchgeführte Transplantationen bei verschiedenen
Indikationen.
Damit stehen den Transplantationszentren wichtige Referenzgrößen zur
Beurteilung, wie auch zur Planung weiterer Aktivitäten und Studien zur
Verfügung. Darüber hinaus unterstützt das DRST die Durchführung von
nationalen und internationalen wissenschaftlichen Studien aktiv.
Es arbeitet eng mit dem europäischen Zentrum (European Group for Blood
and Marrow Transplantation, Maastricht, Niederlande) zusammen. Dort
werden europaweit alle Blutstammzelltransplantationen in einer Datenbank
gespeichert.
nach oben
zur Projektseite
home
12/6
Molekulare
Targets der alternden Mämatopoese bei Patienten mit myelodys-plastischem
Syndrom (MDS)
Prof. Dr.
Wolf-Karsten Hofmann, Direktor der III. Med. Klinik Hämatologie und
Onkologie,
Universitätsmedizin Mannheim, Theodor-Kutzer-Ufer 1 - 3, 68167 Mannheim
Myelodysplastische Syndrome
(MDS) sind eine bösartige Erkrankung des Knochenmarks und resultieren in
einer unzureichenden Produktion von Blutzellen. Insbesondere der Mangel
an roten Blutkörperchen führt zu einer ernsthaften Unterversorgung des
Körpers mit Sauerstoff. MDS treten vorrangig in der älteren
Bevölke-rungspopulation auf und weisen eine dramatische Zunahme der
Inzidenz über 60 Jahre auf. Beim MDS geht man davon aus, dass eine
entartete Stammzelle mehrere Veränderungen im Erbgut erfahren hat und
deshalb für eine übermäßige Produktion gestörter Blutzellen
verantwortlich ist.
Mit der Zunahme des durchschnittlichen
Lebensalters in der deutschen (und europäischen) Bevölkerung hat man in
den letzten Jahren immer wieder festgestellt, dass ältere, zunächst
gesund wirkende Personen, zum Teil ähnliche Symptome wie MDS-Patienten
aufweisen. Häufig wird eine Abnahme der Lymphozytenzahl und damit
Schwächung des Immunsystems oder eine Verringerung der Produktionsrate
von Blutzellen beob-achtet. Neueste Studien liefern Hinweise darauf,
dass nicht nur Schäden in den blutbildenden Stammzellen, sondern auch in
den Vorläuferzellen der roten Blutkörperchen für diese Veränderungen
ursächlich sein können.
In vorliegenden Projekt sollen Stammzellen und Vorläuferzellen der roten
Blutkörperchen aus dem Knochen-mark von gesunden jungen und alten
Probanden sowie MDS Patienten isoliert werden. Im nächsten Schritt
erfolgt eine molekular-genetische Analyse durch eine innovative
Kombination neuester Hochdurchsatz-techniken, die es ermöglicht, die
gesamte DNA auf kleinste Schäden und bestimmte chemische
Modifi-kationen, die zum An- oder auch Abschalten von Genen führen, zu
analysieren. Da somit fast alle Gene auf potentielle Veränderungen
untersucht werden, sollen auf diese Weise Signalwege ermittelt werden,
die zu den oben beschriebenen Veränderungen der Blutbildung im
Alter und bei MDS Patienten führen.
Es wird angestrebt. altersabhängige und MDS-spezifische Veränderungen zu ermitteln und die Hypothese zu überprüfen, ob MDS als natürliche Kontinuität aus der alternden Blutbildung entsteht oder als eigen-ständiger Symptomenkomplex zu betrachten ist.
Veröffentlichung in Experimental Hematology Volume 37, Issue 2 , Pages 215-224.e2, February 2009
nach oben zur Projektseite home
12/7
Aberrante
Chromatin-Modellierung durch das t(6;9) assoziierte Fusionsprotein
DEK/CAN
PD Dr. Martin
Ruthardt, Zentrum für Innere Medizin, Medizinische. Klinik II Abteilung
Hämatologie,
Klinikum der Goethe-Universität, Theodor Stern Kai 7, 60596 Frankfurt am
Main
Die aktuelle myeloische Leukämie (AML) ist eine klonale Erkrankung einer
frühen myeloischen Vorläurger-zelle. Bei zwei Dritteln der AML-Patienten
können numerische oder strukturlle Chromosomenaberrationen nachgewiesen
werden, denen eine ursächlich Rolle in der Leukämogenese zugeschrieben
werden.
Bei den strukturellen Chromosomenaberrationen handelt es sich in den
meisten Fälle um reziproke Chromonentranslokationen.
Einen besonderen Stellenwert nimmt
die t(6;9)-positive AML ein, da sie im Gegensatz zu den anderen AML
Subtypen, die Erkrankungen des Alters darstellen, gehäuft bei jungen
Patienten auftritt und mit einer extrem schlechten Prognose
vergesellschaftet ist. Das hat dazu geführt, dass die t(6;9)-positive
AML in der neuen WHO-Klassifikation der AML des Erwachsenen als eigene
Entität geführt wird. Die t(6;9)(p23q34) kodiert das DEK/CAN
Fusionsprotein, das wie andere AML-assoziierte Fusionsproteine, z.B.
PML/RARα, oder AML-1/ETO ein aberranter Transkriptionsfaktor ist.
Die genetische Information einer
Zelle ist im so genannten Chromatin verpackt, in dem die DNA um
Strukturen, die von den Histonen gebildet werden, gewunden ist.
Veränderungen dieser Strukturen, durch Methylierung, bzw. Azetylierung
steuern die korrekte Verarbeitung der genetischen Information in der
Zelle und erlauben, dass ein Gen in das zugehörige Eiweiß umgeschrieben,
bzw. nicht umgeschrieben wird. Diese Veränderungen an der „Verpackung“
führen dann zu Strukturveränderungen, direkt an der DNA, die auch an
bestimmten Stellen methyliert werden kann. Dadurch wird das zugehörige
Gen abgeschaltet. Diese Prozesse werden unter dem Begriff Epigenetik
zusammengefasst.
Epigenetik bedeutet die Veränderung
der Umsetzung der Erbinformation ohne die in der DNA abgelegten
Erbinformation selbst zu verändern. Leukämie-induzierende Faktoren, wie
die Translokationprodukte PML/RAR oder AML-1/ETO, sind in der Lage, die
Chromatinstruktur aberrant zu modifizieren, so dass es zu ungewünschten
Aktivierungen, bzw. Hemmungen von wichtigen Genen kommen kann, was zur
Leukämieentstehung beitragen kann.
Ziel des hier vorgestellten
Projekts ist es, in einem Modell, das epigenetisch noch kaum
determiniert ist, den so genannten embryonalen Stammzellen der Maus den
Einfluss von DEK/CAN auf die Epigenetik zu untersuchen. Dadurch soll
geklärt werden, welche Bedeutung die epigenetischen Veränderungen für
die durch DEK/CAN induzierte Leukämogenese hat. Damit soll das
Verständnis für die Mechanismen der Leukämogenese erweitert werden und
neue Grundlagen für molekulare Therapieansätze geschaffen werden.
nach oben
zur Projektseite
home
12/8
Identifizierung
klinisch relevanter Adhäsionsmoleküle bei der kindlichen akuten
lymphoblastischen Leukämie
Dr. Leonid Karawajew, Charité Campus Buch, Lindenberger Weg 80, 13122 Berlin
Die akute lymphoblastische Leukämie (ALL) im
Kindesalter ist keine einheitliche Erkrankung sondern umfasst
verschiedene Unterformen, die sich bezüglich Prognose und
Krankheitsverlauf zum Teil deutlich voneinander unterscheiden. Dies
erfordert unterschiedliche Behandlungsstrategien um das Rückfallrisiko
des Patienten zu minimieren.
Für diese risikoadaptierte Behandlung werden die Patienten anhand von
initialen prognostischen Faktoren und Therapieverlaufsparametern in
Risikogruppen stratifiziert. Die Stratifizierung erlaubt eine
Klassifizierung von ca. 50 % der Patienten in Standard- und
Hochrisiko-Gruppe. Aufgrund von fehlenden spezifischen Risikofaktoren
werden die restlichen Patienten als Gruppe des intermediären Risikos
(IR) zusammen-
gefasst. Ihre klinische Bedeutung folgt aus der Tatsache, dass die
Hälfte aller Rezidive aus der IR-Gruppe hervorgeht.
Unsere Vorarbeiten zur Identifizierung von Parametern, die mit dem
unterschiedlichen klinischen Verlauf assoziiert sind, deuten darauf hin,
dass sich die rezidivfreie und rezidivierende ALL in ihren
Wechselwir-kungen mit der Mikroumgebung (dem Stroma) unterscheiden.
Diese Erkenntnisse konnten mittels Xenotransplantaten in Stromabasierten
in vitro Versuchen und in vivo im NOD/SCID-Mausmodell
gewonnen werden. Im beantragten Projekt sollen die Moleküle, die
entscheidend für die Wechselwirkungen von ALL und Mikroumgebung sind,
identifiziert werden.
Hierfür wird ein vorhandenes Analyseverfahren (ein
durchflusszytrometrisches Mikrobead-basiertes Test-system)
weiterentwickelt und an die leukämiespezifische Fragestellung
angepasst. Das Screening potentieller All-Stroma-Interaktionspartner und
die Generierung von Expressionsprofilen für die IR-ALLs mit und ohne
Rezidiv sollen dazu beitragen, das Rezidivrisiko besser vorherzusagen
und zielgerichtete Thera-pien zu entwickeln, die eine optimierte
Risikoadaptierte Behandlung der Patienten ermöglichen.
nach oben
zur Projektseite
home
12/9
Der onkogene Transkriptionsfaktor TAL 1 als
Regulator des alternativen
Splicing; Untersuchungen zum Einfluss von TAL1 auf die
Isoformgenerierung
in Leukämiezellen
Dr.
rer. nat. Jörn Lausen, Gruppenleiter, Georg Speyer Haus,
chemotherapeutisches Forschungsinstitut, Paul-Ehrlich-Str. 42 - 44,
60596 Frankfurt a. M.
Der Transkriptionsfaktor Tal1 ist wichtig für die Entstehung von
Blutstammzellen und für die Ausbildung reifer
Blutzellen wie z. B. rote Blutkörperchen. Tal1 steuer die Ablesung von
Genen während der Entwichlung von Blutzellen. Dies ist wichtig, da ein
zuviel oder zu wenig bestimmter Genprodukte Krankheiten im Menschen
auslösen kann. Wird die Funktion von Tal1 durch Mutationen verändert,
kommt es zu einer Fehlsteuerung der Genablesung und dies kann zr
Leukämie führen.
Einige Gene können alternative Genprodukte kodieren, z. B., kann die
Information eines Gens in ver-schieden lange Proteine übersetzt werden,
die sich in ihrer Funktion unterscheiden. Es ist bekannt, das die
Balance dieser verschiedenen Ioformen wichtig für die normale
Entwicklung von Zellen ist. Wie diee Balance aufrechterhalten und
gesteuert wird und wie sie bei Leukämien gestört ist, ist wenig bekant.
In diesem Forschungsprojekt möchten wir den Einfluss von Tal1 auf die
Entstehung vereschiedener Protein-isoformen untersuchen. Dazu werden wir
Tal1 gezielt in Zellen herabregulieren und die Bildung veränderter
Isofomrn nach der Manipulation von Tal1 genomweit untersuchen. Dazu wird
die neue Methode der Hoch-durchsatzsequenzierung (DeepSequencing)
eingesetztl
Dies erlaubt uns, alle Isoformveränderuongen gleichzeitig und genomwet
zu erfassen. Durch anschließende bioinformatische Analyse wollen wir
zelluläre Mechanismen ausfindig machen, die durch veräderte Isofor-men
betroffen sind. Diese möchen wir weitergehend untersuchen. Wir hoffen,
dass die von uns durchge-führten Untersuchungen bisher unbekannte
Regulationsmechanismen von Tal1 aufdecken werden, die als Ziele für eine
therapeutische Intervention inn Frage kommen.
nach oben
zur Projektseite
home
geförderte Projekte 2013
13/1
Molekulare Targets der alternden
Hämatopoese bei Patienten mit myelodysplas- ischem
Syndrom
Prof. Dr. Wolf-Karsten
Hofmann, Direktor der III. Med. Klinik Hämatologie und Onkologie,
Universitätsmedizin Mannheim, Theodor-Kutzer-Ufer 1 - 3, 68167 Mannheim
Fortsetzungsförderung aus dem Jahr 2012
Myelodysplastische Syndrome (MDS)
sind eine bösartige Erkrankung des Knochenmarks und resultieren in einer
unzureichenden Produktion von Blutzellen. Insbesondere der
Mangel an roten Blutkörperchen führt zu einer ernsthaften
Unterversorgung des Körpers mit Sauerstoff. MDS treten vorrangig in der
älteren Bevölkerungspopulation auf und weisen eine dramatische Zunahme
der Inzidenz über 60 Jahre auf.
Beim MDS geht man davon aus, dass eine entartete Stammzelle mehrere
Veränderungen im Erbgut erfahren hat und deshalb für eine übermäßige
Produktion gestörter Blutzellen verantwortlich ist. Mit der Zunahme des
durchschnittlichen Lebensalters in der deutschen (und europäischen)
Bevölkerung hat man in den letzten Jahren immer wieder festgestellt,
dass ältere, zunächst gesund wirkende Personen, zum Teil ähnliche
Symptome wie MDS-Patienten aufweisen.Häufig wird eine Abnahme der
Lymphozytenzahl und damit Schwächung des Immunsystems oder eine
Verringerung der Produktionsrate von Blutzellen beobach-tet.
Neueste Studien liefern Hinweise darauf, dass
nicht nur Schäden in den blutbildenden Stammzellen, sondern auch in den
Vorläuferzellen der roten Blutkörperchen für diese Veränderungen
ursächlich sein können.
Im vorliegenden Projekt sollen Stammzellen und Vorläuferzellen der roten
Blutkörperchen aus dem Knochen-mark von gesunden jungen und alten
Probanden sowie MDS Patienten isoliert werden. Im nächsten Schritt
erfolgt eine molekulargenetische Analyse durch eine innovative
Kombination neuester Hochdurchsatztechni-ken, die es ermöglicht, die
gesamte DNA auf kleinste Schäden und bestimmte chemische Modifikationen,
die zum An- oder auch Abschalten von Genen führen, zu analysieren. Da
somit fast alle Gene auf potentielle
Veränderungen untersucht werden, sollen auf diese Weise Signalwege
ermittelt werden, die zu den oben beschriebenen Veränderungen der
Blutbildung im Alter und bei MDS Patienten führen.
Es wird angestrebt. altersabhängige und MDS-spezifische Veränderungen zu
ermitteln und die Hypothese zu überprüfen, ob MDS als natürliche
Kontinuität aus der alternden Blutbildung entsteht oder als
eigen-ständiger Symptomenkomplex zu betrachten ist.
nach oben
zur Projektseite
home
13/2
Integrierte Analyse von DNA-Methylierung und
Genexpression bei Patienten mit myelodysplastischem Syndrom und
Deletion am Chromosom 5 q
Prof. Dr.
Wolf-Karsten Hofmann, Direktor der III. Med. Klinik Hämatologie und
Onkologie,
Universitätsmedizin Mannheim, Theodor-Kutzer-Ufer 1 - 3, 68167 Mannheim
Das myelodysplastische Syndrom (MDS) stellt
eine Modellerkrankung der Hämatopoese dar und es konnte bisher durch
verschiedene zellbiologische und molekulargenetische Methoden gezeigt
werden, dass für die leukämische Transformation der hämatopoetischen
Stammzelle die Kumulation einer Vielzahl von endo-genen und exogenen
Ereignissen erforderlich ist.
In Vorarbeiten zur Genexpression und zur DNA-Methylierung in adulten
hämatopoetischen Stammzellen und Zellen in verschiedenen
Differenzierungsstufen der Hämatopoese (sowohl normale Zellen als auch
Zellen von Patienten mit MDS) konnte gezeigt werden, dass es
MDS-spezifische Methylierungsmuster für wichtige, den Zellzyklus und die
Differenzierung kontrollierende Gene gibt.
Im vorliegenden
Projekt sollen diese DNA-Methylierungsmuster und korrespondierende
Genexpressions-muster mit einer arraybasierenden Technik an
Patientenproben einer speziellen Form des MDS (MDS 5q-Syndrom)
untersucht werden. Dabei ist es das Ziel eine biostatistische
Analysemethode zu entwickeln, die die integrierte Datenanalyse erlaubt.
nach oben
zur Projektseite
home
13/3
Dokumentationsprojekt zur Praxis der
Therapie der akuten Graft versus Host Disease an deutschen
Transplantatonszentren
Dr. Daniela Heidereich /
PD Dr. Stefan A. Klein, Universitätsmedizin Mannheim,
III. Med. Klinik Theodor-Kutzer-Ufer 1 -3, 68167 Mannheim
siehe 2011 8
Die Fortsetzung der
Förderung des Projektes erfolgt, da sich herausgestellt hat, dass
eine einjährige Laufzeit nicht ausreicht, um das Projekt erfolgreich zum
Abschluss zu
bringen.
In Kooperation mit dem Deutschen Register
Stammzelltransplantation (DRST) werden
an Hand der Datensätze aus den teilnehmenden Zentren aller Patienten mit
aGvHD mit
AML oder MDS als Grunderkrankung identifiziert.
Es sind mindestens. 500 Patienten eingeschlossen. Das DRST stellt die
Datensätze aller Patienten, die nach der Transplantation eine GvHD
erlitten haben, zur Verfügung.
Aufbauend auf diesen Daten werden vor Ort in den Transplantationszentren
die GvHD-
spezifischen Daten erfasst, so dass einheitliche Datensätze sehr hoher
Qualität
gewonnen werden.
nach oben
zur Projektseite
home
13/4
Unterstützung
des Deutschen Registers für Stammzelltransplantationen DRST
Das Deutsche Register
für Stammzelltransplantationen (DRST), gegründet am
03. 04.1998 in Frankfurt am Main, ist zuständig für die zentrale
Erfassung und
standardisierte Auswertung aller in deutschen Transplantationszentren
nach dem
01.01.1998 durchgeführten Transplantationen und liefert zeitnah wichtige
Daten
über durchgeführte Transplantationen bei verschiedenen
Indikationen.
Damit stehen den
Transplantationszentren wichtige Referenzgrößen zur Beurteilung,
wie auch zur Planung weiterer Aktivitäten und Studien zur Verfügung.
Darüber hinaus
unterstützt das DRST die Durchführung von nationalen und internationalen
wissen-
schaftlichen Studien aktiv.
Es arbeitet eng mit dem europäischen Zentrum (European Group for Blood
and Marrow
Transplantation, Maastricht, Niederlande) zusammen. Dort werden
europaweit alle
Blutstammzelltransplantationen in einer Datenbank gespeichert.
nach oben
zur Projektseite
home
13/5
Einfluss von Hypoxie auf die
Interaktion von Leukämieblasten
mit Knochenmark-Stromazellen
Dr.
rer. nat. Jasmin Wellbrock,
Prof. Dr. Walter Fiedler (Arbeitsgruppenleiter)
Universitätsklinikum Hamburg-Eppendorf, Onkologisches Zentrum,Abteilung Hämatologie und Onkologie
Martinistraße 52,
20246 Hamburg
Die akute myeloische Leukämie (AML) ist eine
maligne Erkrankung des blutbildenden Systems mit äußerst schlechter
Prognose für die Patienten.
Leukämie-Stammzellen (leukemic stem cells, LSC) sind in den Fokus der
zielgerichteten Therapie-Strategien gerückt, da sie aufgrund ihrer
Chemotherapie-Resistenz für die schlechte Prognose der AML
verantwortlich gemacht werden. Im Knochenmark stehen die LSCs innerhalb
der so genannten Leukämie-Stammzell-Nische in enger Interaktion mit
einer Reihe von Stromazellen (umgebende Knochenmarkzellen).
Obwohl bekannt ist, dass das Milieu innerhalb der
Leukämie-Stammzell-Nische nur über einen sehr geringen Sauerstoff-Anteil
verfügt, wurde der Großteil der bisherigen In-vitro- Studien über
die Wechsel-wirkung von LSCs und Stromazellen unter normoxischen
(normalen) Bedingungen durchgeführt. Es gibt jedoch erste Hinweise
darauf, dass die Sauerstoff-Konzentration Einfluss auf die Interaktion
von Leukämie- und Stromazellen nimmt.
Aus diesem Grund möchten wir in dem geplanten Projekt analysieren, wie
sich das Zu-sammenspiel von LSCs und ihren Stromazellen unter
Mangelversorgung mit Sauerstoff (Hypoxie) im Vergleich zu Normoxie
verändert. In früheren Studien konnten wir zeigen, dass sowohl der
vaskuläre Wachstumsfaktor VEGF
(Vascular Endothelial Growth Factor) als auch die stammzellrelevanten
Signalwege (die Hedgehog-sowie die Roundabout-Signalkaskade) eine
wichtige Funktion beim Zusammenwirken von LSCs und ihren Stromazellen
innehaben.
Daher möchten wir den Fokus des geplanten Projektes zunächst auf diese
drei Signalwege legen.Das Ziel des Versuchsvorhabens ist, den Einfluss
von Hypoxie auf die Interaktion von Leukämiezellen mit den sie
umgebenden Knochenmark-Stromazellen zu analysieren.Hierbei soll der
Fokus zunächst auf einige Signalwege gelegt werden, für die wir in
Vorarbeiten nachweisen konnten, dass sie eine wichtige Rolle beim
Zusammenspiel von Leukämie- und Stromazellen übernehmen. Hierzu zählen
neben dem VEGF der Hedgehog- sowie der Roundabout-Signalweg.
nach oben
zur Projektseite
home
13/6
Identifizierung
klinisch relevanter Adhäsionsmoleküle bei der kindlichen akuten
lymphoblastischen Leukämie
Dr.
Leonid Karawajew, Charité Campus Buch, Lindenberger Weg 80, 13122 Berlin
Fortsetzungsförderung
des 2012 begonnenen Projektes
Mitfinanzierung der Stelle einer MTA
In den letzten Jahren tritt zunehmend die Bedeutung der Tumor-Mikroumgebung für die Pathophysiologie der akuten und chronischen Leukämien zutage. Adhäsionsmoleküle sind zentrale Regulatoren der Interaktionen von Tumorzellen mit der Mikroumgebung (Stroma) und stellen somit mögliche zukünftige therapeutische Angriffspunkte in der Behandlung dar.
Unsere Ergebnisse bei der akuten lymphoblastischen Leukämie (ALL) im Kindesalter weisen auf eine wichtige Funktion der Mikroumgebung für die Genese, Progression und Resistenz der Tumorzellen hin. So zeigen die Genexpressionsveränderungen in den ALL- und Stromazellen eine gegenseitige Induktion von Signalmediatoren, die auf ein konzertiertes Zusammenspiel verschiedener Signalwege bei der Kommunikati-on zwischen ALL und Stroma schließen lassen.
Um dieses Ergebnis auf Proteinebene zu validieren wurde ein durchflusszytometrisches Mikrobead-basiertes Testsystem etabliert. Im Rahmen des vorliegenden Projektes ist es geplant Adhäsionsprofile von 30 bis 50 Knochenmarkproben von ALL-Patienten bei Diagnosestellung zu generieren. Klinisch relevante Proteine sollen anschließend prospektiv an größeren Patientenkohorten evaluiert werden. Ein systematisches Screening potentieller ALLStroma-Interaktionspartner soll zur Identifizierung von funktionellen Rezeptoren führen, die an den Wechselwirkungen von Leukämie- und Stromazellen beteiligt sind.
Die Generierung von ALL-spezifischen Interaktionsprofilen soll dazu beitragen, das Rezidivrisiko besser vorherzusagen und zielgerichtete Therapien zu entwickeln, die eine optimierte Risiko-adaptierte Behandlung der Patienten ermöglichen.
nach oben
zur Projektseite
home
13/7
Neue translationale Bioinformatikmethoden zur integrativen
Analyse multidimensi-onaler Patientendaten aus traditionellen und high
throughput Quellen
Ralf Bieber M. Sc. Systemarchitekt und Bioinformatiker; Prof. Dr. med. Wolf-K. Hofmann, Universitäts-medizin Mannheim, III. Med. Universitätsklinik, Hämatologie und Onkologie - Wissenschaftliches Labor, Pettenkofer Str. 22, D-68169 Mannheim
(Definition aus Wikipedia)
Translationale Medizin
ist die Schnittstelle zwischen präklinischer Forschung und klinischer
Entwicklung. Sie beschäftigt sich mit der Übersetzung von z. B.
in-vitro- Modellen oder Tiermodellen in die Anwendung am Menschen.
In der biologischen Forschung steht der
Begriff High Throughput für eine Klasse von Technologien die Dank
parallelisierter Anordnung einzelner Analysereaktionen, eine große
Bandbreite von Molekülen zur gleichen Zeit, auf eine Fragestellung
analysieren können. Die am weitesten verbreiteten Technologien dieser
Gruppe sind die Massenspektrometrie, Microarrays und die
Sequenziertechnologien der zweiten Generation (Next Generation
Sequencing - NGS).
Nach
aktuellem Stand der Technologie ist die gesamte Rechen- und
Speicherleistung von Forschung und Industrie nicht ausreichend, um die
täglich produzierten Sequenzierungsdaten aus "Next Generation Sequencing
(NGS)"-Maschinen zeitnah auszuwerten oder zu speichern. NGS ist nur ein
Hochdurchsatzver-fahren das Wissenschaftler, neben klassischen Methoden,
zur Verfügung steht. Dies führt zum aktuell gültigen Paradigma: ‚Eine
Datenquelle, eine Auswertung mit Wochen an Berechnungszeit‘. Das
zusätzliche Verbinden der Datenquellen und funktionale Verknüpfen der
Ergebnisse hat bis dato eine Komplexität, der sich nur wenige
interdisziplinäre Arbeitsgruppen gestellt haben.
Das Verarbeiten der
Daten benötigt immer öfter mehr Zeit als die meisten anderen Schritte in
einem Forschungsprojekt und ist nur von gut ausge-bildeten Spezialisten
ausführbar. Das Schaffen von
Methoden zur Verknüpfung der Daten zwischen techno-logischen Grenzen ist
eine richtungsweisende Aufgabe der translationalen Bioinformatik.
Um die stetig wachsenden Anforderungen an
Datenhaltung und Auswertung eines Labors zukunftssicher abzudecken,
wurde von unserer Abteilung ein eigens dafür konzipiertes
Informationssystem entwickelt. Die Software ist zudem für die
Darstellung und Verwaltung verschiedenster Laborprozesse optimiert. Die
zu-grundeliegende Technologie des Informationssystems beruht auf
aktuellen und offenen Technologien ("open-source") sowie modernen
Architekturmustern. Unter Berücksichtigung der stetig wachsenden
Datenmengen der Medizin und Biologie wurde eine Architektur
implementiert, die eine vereinfachte und kostengünstige Adaption neuer
Anforderungen und Problemstellungen ermöglicht.
In projektorientierter Weise werden bereits heute Routine- wie auch Forschungsdaten verarbeitet und strukturiert gespeichert. Eine Kooperation mit der Hochschule Mannheim – Fachbereich Medizinische Informatik ist ein Bezugspunkt für aktuelles und hochwertiges Know How, das in weiteren Projekten der Grundlagenforschung direkt zur Anwendung kommt.
Ziel der Förderung ist die Erweiterung des Informationssystems um standardisierte, wiederverwendbare Architekturen und Methoden zur Unterstützung der Routinediagnostik und Forschung, der Ausbau der Infrastruktur zur Beschleunigung der Auswertung, bzw. Ausgleich des Wachstums der Datenmengen und die Ausbildung und Förderung von Nachwuchswissenschaftlern im Studium der Medizin- / Bio- Informatik durch Ausschreibung von Stellen für Studentische Wissenschaftliche Hilfskräfte zum Schaffen von Kompetenz und Know How während des Studiums.
nach oben zur Projektseite home
13/8
Genomische Architektur der akuten
lymphoblastischen Leukämie und deren klonale Evolution im Rezidiv
Prof. Dr. med. Claudia Baldus / Lorenz Bastian, Charité, Campus Benjamin
Franklin
Hämatologie, Onkologie, Tumorimmunologie, Hindenburgdamm 30, 12203
Berlin
Die
Prognose der rezidivierten akuten lymphoblastischen Leukämie (ALL) im
Erwachsenenalter ist alles andere als zufriedenstellend. Die Ursachen
des im Vergleich zur ALL im Kindesalter deutlich häufigeren Auftretens
von Rezidiven und des sehr viel schlechteren Therapie-Ansprechens
sind allenfalls anfänglich verstanden. Neben einer schlechteren
Verträglichkeit von dosisintensiven konventionellen Therapien scheinen
vor allem genetische Erkrankung bei Erwachsenen zu bestimmen.
Mit der Identifizierung
der BCR/ABL-Translokation
konnte für eine Subgruppe von 25 % der Patienten ein genetischer Treiber
der Erkrankung mit einem besonders aggressiven Verlauf gefunden werden.
Für die übrigen Patienten stehen zum Teil genetische Marker zur
Verfügung, die auf eine ungünstige Prognose hinweisen. Diese genetischen
Veränderungen sind jedoch nicht hinreichend, um als ursächlich für die
Krankheitsentstehung zu gelten und als therapeutische Zielstrukturen zu
fungieren. Zudem finden sich in
23 % der Patienten mit den etablierten zyto-
und molekulargenetischen Methoden bisher keine molekularen
Veränderungen.
Derzeit ist unklar, wie sich die genetischen Treiber der
Krankheitsbiologie von der Erstdiagnose zum Rezidiv verhalten. Es ist
nicht bekannt, ob sich das Rezidiv aus primär resistenten Zellen speist,
ob unter dem Einfluss der Chemotherapie resistente Subpopulationen von
Leukämiezellen selektiert werden, oder ob gänzlich neue Subpopulationen
erst entstehen.
Die neue Methode des Next Generation Sequencing, die wir im Rahmen von
erfolgreich abgeschlossenen Projekten bereits mehrfach eingesetzt haben,
erlaubt eine hochauflösende Charakterisierung von geneti-schen
Veränderungen in Leukämiezellen.
Im Rahmen des
beantragten Projekts soll anhand von 556 biologisch relevanten
Kandidaten-Genen
bei 40 Patienten das genomische Profil der ALL des Erwachsenenalters
charakterisiert werden.
Durch die Untersuchung von diagnostischem Material aus Erstdiagnose und
Rezidiv für jeden dieser Patien-ten und den Abgleich mit klinischen
Daten des Krankheitsverlaufs sollen einerseits potentielle genetische
Treiber identifiziert und andererseits deren Entwicklung von der
Ersterkrankung zum Rezidiv (klonale Evolution) analysiert werden.
Durch Zuordnung der gefundenen genetischen Veränderungen zu einzelnen
Signalwegen sollen Angriffs-punkte für neue molekulare Inhibitoren
identifiziert werden, von denen bereits eine Vielzahl für
unterschied-liche Signalwege zur Verfügung steht. Basierend auf dem
genetischen Hintergrund der Krankheitsbiologie ergeben sich damit neue
Perspektiven für eine zielgerichtete und individualisierte Therapie der
ALL im Erwachsenenalter.
nach oben
zur Projektseite
home
geförderte Projekte 2014
14/1
Analyse genetische Veränderungen in mesenchymalen Stromazellen von
Patienten mit myelodysplastischem Syndrom (MDS) in einem neuen
in-vivo-Modell
Prof. Dr. med.
Wolf-Karsten Hofmann, Direktor der III. Med. Klinik Hämatologie und
Onkologie,
PD Dr. med. Daniel Nowak, III. Med. Klinik Hämatologie und Onkologie,
Universitätsmedizin Mannheim, Theodor-Kutzer-Ufer 1 - 3, 68167 Mannheim
Vorbemerkung zum
myelodysplastischen Syndrom (MDS)
Das myelodysplastische Syndrom (MDS) ist eine klonale maligne Erkrankung der Hämatopoese. Das Krankheitsbild ist gekennzeichnet durch eine periphere Zytopenie, hier insbesondere führend eine Anämie. Die Erkrankung betrifft besonders Patienten in fortgeschrittenem Alter, das mediane Alter bei Erstdiagnose beträgt ca. 70 Jahre. Die Standardbehandlung des MDS besteht in der Transfusion von Erythrozytenkonzentraten, eine Vielzahl von experimentellen Therapien von Wachstumsfaktoren über demethylierende Substanzen bis hin zu immunmodulatorischen Medikamenten kommen zur Anwendung, um das Ausmaß der hämatopoetischen Insuffizienz zu verringern. Die einzige kurative Therapieoption für Patienten mit MDS besteht zurzeit in der allogenen Stammzell-transplantation.
Ein großes Problem und ein die Prognose bestimmender Umstand ist, dass die frühe Form des myelodys-plastischen Syndroms, die in der Regel nur durch eine Anämie, Bi- oder Trizytopenie gekennzeichnet ist, sich im Verlauf der Erkrankung verändern kann und es zu einer Entwicklung einer akuten myeloischen Leukämie kommt. Durchschnittlich 30-50% der Patienten erleiden eine solche Transformation innerhalb von 3 Jahren. Es handelt sich dann in der Regel um eine Hochrisiko-Leukämie, die therapeutischen Optionen sind bei den älteren Patienten für diese Erkrankung sehr eingeschränkt.
Die Pathogenese des MDS ist bis heute nicht geklärt. Sieht man von den wenigen Fällen sogenannter sekundärer MDS ab, welche durch die langanhaltende Exposition des betroffenen Patienten zu benzol-haltigen chemischen Stoffen bedingt sein können, kann in über 95 % der Fälle eine Ursache der Erkrankung nicht festgestellt werden. Zwei grundlegende Theorien für die Entstehung der primären Formen des MDS werden heute angenommen:
1. Modell der kompletten klonalen Hämatopoese. Ähnlich wie bei der akuten Leukämie wird davon ausge-gangen, dass das Knochenmark komplett von Zellen eines malignen Klones eingenommen wird. Aus noch ungeklärten Gründen besteht weiterhin eine teilweise Blutbildung, die vorhandenen reifen hämatopoetischen Zellen weisen jedoch strukturelle und funktionelle Defekte auf. Nach einer bis zu mehreren Jahren dauernden „chronischen Phase“ kommt es im Verlauf zur Transformation der Erkrankung zur akuten Leukämie.
2. Modell der Expansion eines malignen Klones neben normaler Hämatopoese. Bei diesem Modell nimmt man an, dass sich Zellen des malignen Klones allmählich neben der normalen Blutbildung etablieren. Es kommt zu einer zunehmenden Verdrängung der normalen Hämatopoese, gleichzeitig wird die Proliferation und Differenzierung der polyklonalen Zellen durch inhibitorische Zytokine (Tumor Nekrose Faktor alpha, TNFa) gehemmt. Die bestehende Restblutbildung kann über Jahre hinweg funktionieren, bis es zur Expan-sion des malignen Klones und damit zur Verdrängung der normalen Blutbildung kommt.
Zusammenfassung
Beim
myelodysplastischen Syndrom (MDS) handelt es sich um eine
Modellerkrankung der Hämatopoese. Durch verschiedene zellbiologische und
molekulargenetische Methoden konnte gezeigt werden, dass für die
leukämische Transformation der hämatopoetischen Stammzelle die
Kumulation einer Vielzahl von endo-genen und exogenen Ereignissen
erforderlich ist. In einem kürzlich von der Arbeitsgruppe Nowak/Hofmann
in Kooperation mit dem Deutschen Krebs Forschungs Zentrum (DKFZ,
Arbeitsgruppe Trumpp) etablierten
in-vivo-Modell für das MDS konnte erstmals das Anwachsen typischer
MDS-Hämatopoese erreicht werden. Im vorliegenden Projekt sollen
genetische Veränderungen in hämatopoetischen als auch in mesenchymalen
Stromazellen von MDS-Patienten, welche in das in-vivo-Modell
transplantiert worden sind, untersucht werden.
Ziel der Arbeiten für dieses Projekt ist es, die (gestörte?) Interaktion zwischen blutbildenden Zellen und Knochenmarkbindegewebe genau zu analysieren. Damit könnten grundlegende neue Erkenntnisse über die Pathophysiologie des MDS gewonnen werden.
s. a. MDS-Forum 2014
nach oben zur Projektseite home
14/2
Die Rolle der gestörten RNA Prozessierung und
Translation in der durch DEK/CAN getriebenen Leukämogenese bei der
t(6;9)-positiven AML
PD Dr. Martin
Ruthardt, Zentrum für Innere Medizin, Medizinische. Klinik II Abteilung
Hämatologie,
Klinikum der Goethe-Universität, Theodor Stern Kai 7, 60596 Frankfurt am
Main
Das Dogma der Molekularbiologie geht von der Umschreibung der in der DNA
kodierten genetischen Information in “messenger” RNA (mRNA), die
anschließend in den Ribosomen in Proteine übersetzt wird, aus. Aus dem
“human genom project” ging nun hervor, dass der größte Teil der von der
DNA umgeschriebenen mRNA gar nicht in Proteine übersetzt wird. Es ist
weitgehend unklar, welche Rolle diese umgeschriebene aber nicht
übersetzte genetische Information für die Regulierung von biologischen
Prozessen in der Zelle spielen. Immer mehr Daten deuten jedoch darauf
hin, dass sowohl der zelluläre Apparat, der für die Prozessierung von
RNA verant-wortlich ist, sowie die nicht übersetzten RNA-Formen und auch
die Ribosomen selbst, eine bedeutende Rolle in der Regulierung
biologischer Prozesse in der Zelle und bei Erkrankungen wie Krebs
spielen könnten.
Die akute myeloische Leukämie (AML) ist eine klonale
Stammzellerkrankung, bei der die Ausreifung in funktionelle Blutzellen
gestört ist. So kommt es zu einer massiven Anreicherung proliferierender
hämo-poetischer Vorläufer im Knochenmark und im peripheren Blut, die
wiederum die hämopoetische Insuffizienz mit Granulozytopenie, Anämie und
Thrombopenie zur Folge hat.
Spezifische
Translokationen, wie z.B. t(15;17), t(6;9), oder t(8;21) und deren
Genprodukte - PML/RARα, DEK/CAN, und AML-1/ETO - werden für die
Induktion der Leukämie verantwortlich gemacht. In der AML stellt die
t(6;9)-positive AML eine eigene Untergruppe dar. Sie tritt, im Gegensatz
zu den anderen AML Subtypen, gehäuft bei jungen Patienten auf und ist
mit einer extrem schlechten Prognose vergesellschaftet. Das t(6;9)
kodierte DEK/CAN Fusions-protein, besitzt eigenes leukämogenes
Potential, aber der Mechanismus durch den DEK/CAN Leukämie induziert,
ist unbekannt. DEK/CAN benutzt jedoch keinen der Mechanismen anderer
Leukämie-induzieren-der Fusionsproteine.
Mit Hilfe der neuartigen Technik der Massenspektrometrie konnten wir zeigen, dass DEK/CAN sehr eng sowohl mit Proteinen des RNA-Prozessierungsapparats als auch mit Komponenten der Risbosomen assoziiert ist. Diese Daten sind in Übereinstimmung mit neuen Daten einer spanischen Gruppe, die zeigen, dass Patienten mit t(6;9)-positiver AML ein besonderes Muster an sog. microRNAs tragen, das sich von dem anderer AML-Patienten unterscheidet. Diese microRNAs gehören zu den RNAs, die nicht in Proteine übersetzt werden, aber bedeutungsvoll für die Steuerung sehr vieler zellulärer Prozesse sind.
Auf diesen Daten basiert unsere Arbeitshypothese, dass DEK/CAN über einen neuartigen Mechanismus Leukämie induziert, der auf einer Deregulierung sowohl der RNA-Prozessierung als auch seiner Übersetzung in Proteine an den Ribosomen basiert. Aus diesem Grund möchten wir die Frage beantworten, welche Folgen die Interaktion zwischen DEK/CAN und den RNA-bindenden Proteinen für die Prozessierung von RNA, die Expression microRNA, sowie anderer Formen von RNA, die zunehmend Bedeutung für die Steuerung biologischer Prozesse in der Zelle und in Krankheiten gewinnen, haben.
nach oben
zur Projektseite
home
14/3
Molekulare
Charakterisierung der Chromosomentranslokation t(8;22)(q24;q11)
mit MYC-IGL-Juxtaposition bei der ALL vom Burkitt-Typ
PD Dr. med. Dr.
rer. nat. Thomas Burmeister, Facharzt für Innere Medizin/Hämatologie,
Onkologie Charité CBF, Medizinische Klinik für Hämatologie/Onkologie,
12200 Berlin
Hindenburgdamm 30
Das auf Chromosom 8 liegende MYC-Gen spielt eine zentrale Rolle
bei der Entstehung einer großen Zahl von menschlichen Tumorerkrankungen.
MYC beeinflusst eine Vielzahl von anderen Genen. Wird die
Funktion von MYC gestört kann es dadurch zu einer schwerwiegenden
Störung einer Vielzahl von Zell-funktionen kommen und die betroffene
Zelle kann sich zu einer Tumorzelle weiterentwickeln.
Bei einer Untergruppe von bösartigen Erkrankungen des lymphatischen
Systems (malignen Lymphomen, insbesondere Burkitt-Lymphom und
Diffus-großzelliges B-Zell-Lymphom) ist MYC an
Chromosomentrans-lokationen beteiligt. Durch diese
Chromosomentranslokationen gerät das MYC-Gen unter die Kontrolle
anderer Gene und wird in seiner Expression hochreguliert, d. h. das Gen
wird gewissermaßen dauerhaft angeschaltet, mit den oben beschriebenen
Folgen für die betroffene Zelle.
Es handelt es sich im wesentlichen um drei Chromosomentranslokationen,
an denen die Chromosomen 2, 14 oder 22 beteiligt sind, auf denen die
Immunglobulin-Gene liegen.
(Unter einer Translokation (Ortsveränderung, Versetzung, von lateinisch locus: Ort) versteht man in der Genetik eine Chromosomenmutation, bei der Chromosomenabschnitte an eine andere Position innerhalb des Chromosomenbestandes verlagert wurden. Im Extremfall kann sich ein ganzes Chromosom an ein anderes anlagern. Die Translokation wird bei den Chromosomeneigenschaften mit „t“ abgekürzt.)
Ziel des Projektes ist die molekulare
Charakterisierung der Chromosomentranslokation t(8;22)(q24;q11), durch
die das MYC-Gen von Chromosom 8 in die Nähe des IGL
(Immunglobulin-Gen lambda) - Gens gelangt (siehe Abbildung). Diese
genetische Veränderung ist bisher unter mechanistischen Gesichtspunkten
wenig verstanden und auf molekularer Ebene kaum charakterisiert. Es
sollen die genauen Chromosomenbruch-punkte einer größeren Zahl von
Patienten molekular identifiziert werden.
Dadurch soll ein vertieftes Verständnis für das Zustandekommen dieser
genetischen Veränderung gewonnen werden. Da sich
Chromosomentranslokationen mit Beteiligung des IGL-Gens auch bei
vielen anderen Lymphomen finden, sind die hier zu gewinnenden
Erkenntnisse auch für das Verständnis dieser Veränderungen von Relevanz.
Insgesamt soll ein besseres molekulares Verständnis der zugrundeliegenden Erkran-kungen erreicht werden, was letztlich Grundlage für die Entwicklung von neueren und besseren Formen der Diagnostik und Therapie ist.
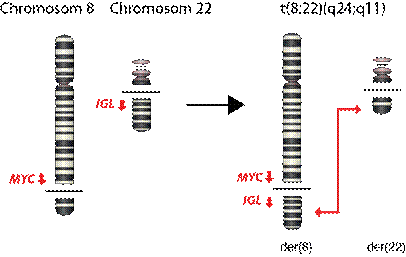
Durch die Chromosomentranslokation
t(8;22)(q24;q11) gelangt das MYC-Gen in die Nachb-arschaft des
IGL-Gens und wird dadurch in seiner Regulation gestört.
nach oben
zur Projektseite
home
14/4
Postranslationale Modifikationen des Onkoproteins Tal 1 als potentielle
molekulare Ziele bei Leukämien
Dr. rer. nat. Jörn Lausen, Gruppenleiter, Georg Speyer Haus, chemotherapeutisches Forschungsinstitut, Paul-Ehrlich-Str. 42 - 44, 60596 Frankfurt a. M.
Der hämatopoetische Transkripionsfaktor Tal1 ist ein wichtiger Regulator
der Genex-pression in hämatopoe-tischen Stammzellen und während der
Differenzierung ver-schiedenartiger Blutzellen. Außerdem ist Tal1 ein
Onkogen bei Leukämien.
In diesen Fällen ist Tal1 aufgrund verschiedener
Mechanismen falsch exprimiert, das heißt, es ist zu viel Tal1 Protein
in der Zelle vorhanden.
Die Tal1-Fehlexpression trägt hier maßgeblich zur Leukämieentstehung
bei, indem Tal1 mit der Wirkung anderer Transkriptionsfaktoren
interferiert. Das Wachstum Tal1-exprimierender Krebszellen ist daher
häufig vom Vorhandensein von Tal1 abhängig.
Eine pharmazeutische Beeinflussung der Tal1-Aktivität könnte daher
anti-proliferativ wirken und könnte ein gangbarer therapeutischer Weg
sein.
Die Aktivität von Tal1 in der Zelle ist durch
posttranslationale Modifikationen stark kontrolliert. Diese
Modifikationen werden durch Enzyme durchgeführt, die chemische Gruppen
an spezifische Aminosäuren von Tal1 anbringen. Dadurch wird die Funktion
von Tal1 als Regulator der Genaktivität verändert. Bei Tal1 ist bisher
vor allem Phosphorylierung beschrieben worden. Wir wollen in diesem
Projekt untersuchen, wie Modifikationen von Tal1 auf die Funktion des
Onkoproteins wirken. Insbesondere sind wir daran interessiert, ob es zu
einer Wechselwirkung verschiedener Modifikationen kommt.
Da die Modifikationen von Tal1 die Funktion des Transkriptionsfaktors
verändern, sind diese für eine therapeutische Beeinflussung des
onkogenen Potentials von Tal1 besonders interessant.
Wir
wollen daher untersuchen, ob Inhibitoren, die bestimmte Modifikationen
von Tal1 beeinflussen, als therapeutische Moleküle bei Leukämien in
Frage kommen.
(Definition aus Wikipedia)
Translationale Medizin ist die Schnittstelle zwischen präklinischer
Forschung und klinischer Entwicklung. Sie beschäftigt sich mit der
Übersetzung von z. B. in-vitro-Modellen oder Tiermodellen
in die Anwendung am Menschen.
nach oben
zur Projektseite
home
14/5
Identizierung von genetischen Faktoren für die Besiedlung und Infektion durch multiseidente Baktereien bfei allogen Stammzelltransplantier
PD Dr. Stefan A.
Klein, Universitätsmedizin Mannheim, III. Med. Klinik,
Theodor-Kutzer-Ufer 1 -3,
68167 Mannheim
Multiresistente Bakterien, insbesondere VRE (Vancomycin reistente
Enterokokken), ESBL
(gram negative Erreger mit Extended Spectrum β-Lactamasen) und
totiresistente Pseudomonaden (totalresistent, gegen die keine
Antibiotika helfen) stellen für schwerst immun-supprimierte Patienten
eine zunehmende Gefahr dar.
Ein wachsender Anteil der Patienten auf hämatologischen Stationen,
insbesondere auch im Bereich der allogenen Stammzelltransplantation ist
mit multiresistenten Erregern besiedelt.
Entwickelt sich aus der Besiedelung eine Infektion, endet diese für die
betroffenen allogen Stammzell-transplantierten Patienten häufig letal.
Zahlreiche Patienten kommen bereits mit einer Besiedlung des Darmes
durch die multiresidenten Erreger
in die Klinik! Bei anderen werden die resistenten Erreger durch den
vielfach unvermeidbaren Einsatz von Breitspektrumantibiotika selektiert.
Trotz vergleichbarer langzeitiger Therapie mit
Antibiotika, wie Carbapenemen, Piperacillin/Tazobaktam oder Vancomycin
werden nur bei einem Teil der Patienten multiresistente Erreger
identifiziert. Darüber hinaus erkrankt von den besiedelten auch nur
wieder ein Teil der Patienten tatsächlich an den resistenten Erregern.
Bislang ist unklar, welche Wirtsfaktoren eine Besiedlung bzw. eine
Infektion begünstigen.
Bei anderen Infektionserkrankungen, wie z. B. Schimmelpilzinfektionen
durch Aspergillus konnten in der Vergangenheit unterschiedliche
single-nucleotide polymorhismus (SNPs) von Spendern und Empfängern für
die Entwicklung von invasiven Schimmelpilzinfektionen identifiziert
werden.
Seit 2011 wird bei sämtlichen Patienten, die auf
die Station für Stammzelltransplantation und Leukämie-therapie der III.
Med. Klinik der Universitätsmedizin Mannheim aufgenommen werden, ein
Screening auf VRE, ESBL, MRSA und multiresistente Pseudomonaden
durchgeführt. Bei ca. 20 % der Patienten können multiresistente Erreger
nachgewiesen werden. Insbesondere bei den mit totiresistenten
Pseudomonaden besiedelten Patienten kommt es unter zytostatischen bzw.
immunsuppressiven Therapien zu schwersten
lebensbedrohlichen Infektionen. Neben Blutstrominfektionen stellen
besonders ausgedehnte Weichteil-infektionen ein großes klinisches
Problem dar.
Patienten, die eine allogene
Stammzelltransplatation erhalten haben und mit einem der oben genannten
Erreger besiedelt oder infiziert sind, werden auf SNPs untersucht.
Hierbei soll sowohl die DNA des Patienten, wie auch des Spenders als
Untersuchungsmaterial herangezogen werden.
Es soll versucht werden, typische rekurrente SNPs (wiederkehrende SNPs)
bei besiedelten und insbe-sondere infizierten Patienten zu
identifizieren.
Es ist die Frage zu klären, ob genetische Wirtsfaktoren bei Spendern und Empfängern im Sinne von SNPs existieren, die bei Patienten unter allogener Stammzelltransplantation für die Besiedlung bzw.Infektion durch multiresistente Erreger prädisponieren.
nach oben zur Projektseite home14/6
Unterstützung des Deutschen Registers für Stammzelltransplantationen DRST
Das Deutsche
Register für Stammzelltransplantationen (DRST), gegründet am 03. 04.1998
in Frankfurt am Main, ist zuständig für die zentrale Erfassung und
standardisierte Auswertung aller in deutschen Trans-plantationszentren
nach dem 01.01.1998 durchgeführten Transplantationen und liefert zeitnah
wichtige Daten
über durchgeführte Transplantationen bei verschiedenen
Indikationen.
Damit stehen den Transplantationszentren
wichtige Referenzgrößen zur Beurteilung, wie auch zur Planung weiterer
Aktivitäten und Studien zur Verfügung. Darüber hinaus unterstützt das
DRST die Durchführung von nationalen und internationalen
wissenschaftlichen Studien aktiv.
Es arbeitet eng mit dem europäischen Zentrum (European Group for Blood
and Marrow Transplantation, Maastricht, Niederlande) zusammen. Dort
werden europaweit alle Blutstammzelltransplantationen in einer Datenbank
gespeichert
14/7
Bedeutung von natürlichen Killerzellen in der erfolgreichen Behandlung des Multiplen Myeloms
Prof. Dr. med. Evelyn Ullrich / Dr. Sabine Hünecke, Universitätsmedizin Klinik für Kinder- und Jugendmedizin, Klinikum der Goethe Universität, Theodoer Stern Kai 7, 60596 Ffm.
In der heutigen Therapie von hoch-malignen Lymphomen und dem Multiplen
Myelom nimmt die Hochdosis-Chemotherapie (HD) mit anschließender
autologer Stammzelltransplantation (autoSZT) eine wichtige Rolle ein.
Durch die HD-Therapie ist eine systematische Elimination maligner Zellen
möglich, doch führt sie zu einer schweren Schädigung des Knochenmarkes.
Durch die anschließende Infusion von autologen Stammzellen kann die
Regenerationsphase im Anschluss an die HD-Therapie verkürzt werden. Ohne
eine solche autoSZT würde die hämatologische Regenerationsphase Wochen
bis Monate dauern, bei gleichzeitig erhöhter Infekt-anfälligkeit.
Nach autoSZT erfolgt die Regeneration der einzelnen Immunzellen zu unterschiedlichen Zeitpunkten. So erfolgt die vollständige Regeneration der B-Zellen erst nach einigen Monaten. Hingegen regenerieren andere Zellreihen, wie z. B. die Natürlichen Killer (NK) Zellen innerhalb weniger Wochen.
NK-Zellen sind wichtige Komponenten der angeborenen Immunabwehr. Ihre Aufgabe besteht in der Kontrolle und Eliminierung von Virus-infizierten oder maligne entarteten Zellen. Ferner können sie andere Immunzellen via Zytokinsekretion beeinflussen. Insbesondere im Zusammenhang mit malignen myeloischen Erkrankun-gen /AML, CML) sowie dem Multiplen Myelom wurde in verschiedenen Phase I/II Studien zum adoptiven Transfer sowohl autologen wie auch allogenen NK-Zellen eine bedeutende antitumorale Funktion zugeschrie-ben. Darüber hinaus wurde bei Patienten mit Multiplem Myelom beobachtet, dass eine erhöhte Zahl von NK-Zellen mit einer günstigeren Prognose des Krankheitsverlaufes korreliert. Allerdings wurde bisher nicht untersucht, welche Bedeutung die Funktionalität der nach SZT regenerierten NK-Zellen für das rezidivfreie Überleben hat.
In diesem Forschungsprojekt sollen daher
erstmals die funktionellen Eigenschaften der NK-Zellen (Lyseaktivität
und Zytokinproduktion) sowie deren Aktivierungsmarker im Verlauf der
HD/autoSZT analysiert werden.
Dabei soll insbesondere eine Analyse der NK-Zellen unmittelbar nach
Regeneration der Leukozyten (<1000/µl) erfolgen, um zu klären, ob
NK-Zellen bereits zu einem sehr frühen Zeitpunkt nach autoSZT
funktionell aktiv sind. Anschließend sollen die in vitro
gewonnenen Daten eines Patienten mit seinem klinischen Verlauf
verglichen und nach möglichen Korrelationen bzw. prädiktiven Markern für
den klinischen Verlauf gesucht werden.
Bericht an die „Alfred- und Angelika Gutermuth-Stiftung“ für das seit 2014 geförderte Projekt:
Bedeutung von Natürlichen Killerzellen in der erfolgreichen Behandlung des Multiplen Myeloms
Projektleitung:
Prof. Dr. med. Evelyn Ullrich (Internistin)
LOEWE
CGT / Klinik für Kinder- und
Jugendmedizin Ffm.
Zusammenfassung der Erebnisse
In der heutigen Therapie mancher Arten des Lymphkonten- und Knochenmarkkrebses, wie z.B. des hoch-malignen Lymphoms und Multiplen Myeloms (auch Plasmozytom genannt), nimmt die Hochdosischemo-therapie (HD) mit anschließender Stammzelltransplantation Patienten-eigener Stammzellen, sog. autologe SZT, eine wichtige Rolle ein. Durch die HD-Therapie ist eine Elimination maligner Zellen möglich, jedoch führt sie zu einer schweren Schädigung des Knochenmarkes. Durch die anschließende Infusion der auto-logen Stammzellen kann die Regenerationsphase im Anschluss an die HD-Therapie verkürzt werden. Die Familie der Natürlichen Killer (NK) Zellen ist eine der ersten Zellpopulationen, die nach SZT regeneriert. Bisher war es jedoch nicht bekannt, ob und in welcher Weise die aus dem Transplantat neu gebildeten NK-Zellen funktionsfähig sind.
In diesem von der Gutermuth-Stiftung unterstützten Forschungsprojekt wurden erstmals die funktionellen Eigenschaften der NK-Zellen sowie deren Aktivierungsmarker zu unterschiedlichen Zeitpunkten vor und nach HD/SZT analysiert. Interessanterweise zeigten diese Untersuchungen, dass schon die ersten neu gebildeten NK-Zellen zu einem sehr frühen Zeitpunkt nach SZT das Potential haben, Tumorzellen zu lysieren und Zytokine (IFN-y und MIP-1ß) zu produzieren. Die hier erhobenen Daten wurden im November 2015 in der international anerkannten Zeitschrift „Frontiers in Immunology“ veröffentlicht (Jacobs et al., Front. Immunol. 2015).
Ergänzend untersucht die Arbeitsgruppe um Prof. Ullrich derzeit, ob aus dem Knochenmark von Patienten gewonnene Multiple Myelom-Zellen von Patienteneigenen, naiven NK-Zellen attackiert und lysiert werden können oder ob es einer ex vivo Aktivierung dieser NK-Zellen bedarf, um eine effiziente Kontrolle der malignen Zellen zu erzielen.
nach oben zur Projektseite home
14/8
Detektion und klinische Bedeutung sehr frühere B-Vorläuferzellen mit aberrantem Immunphänotyp bei Kindern mit akuter lymphoblastischer Leukämie
Dr. rer. nat. Leonid Karawajew, Klinik für
Pädiatrie m.S. Onkologie/Hämatologie, Charité - Universitäts-medizin
Berlin, Lindenberger Weg 80, 13125 Berlin
Die Entstehung von Leukämien lässt sich auf
genetische Aberrationen von hämato-poetischen Stammzellen und
Progenitorzellen zurückführen. In vielen Fällen geht dies mit der
Ausbildung einer Population von leukämischen Zellen mit
Stammzelleigenschaften (leukemia stem cells, LSCs) einher. Obwohl noch
Gegenstand aktueller Diskussion, ist es wahrscheinlich, dass LSCs eine
entscheidende Rolle bezüglich des Wiederauftretens der Erkrankung
spielen.
Es ist somit von großem Interesse, diese Zellen zu identifizieren und
den möglichen Zusammenhang zwischen LSCs und Prognose zu untersuchen.
Es gibt keine eindeutige immunphänotypische Definition der LSCs, es wird
aber angenommen, dass die CD34+CD38- Zellfraktion im Knochenmark mit
LSCs angereichert werden soll. Darüber hinaus wird erwartet, dass die
LSCs keine Differenzierungsmarker, wie CD19 für die B-Zellreihe,
exprimieren und somit den Immunphänotyp CD34+CD38-CD19- aufweisen. Die
Rolle der B-Zellmarker bei den LSCs in ALL der B-Zellreihe (BCP-ALL) ist
aktuell jedoch umstritten.
In der aktuellen Studie zur Behandlung von Kindern und Jugendlichen mit ALL-Rezidiv IntReAll 2010 wurde die multiparametrische Durchflusszytometrie (FCM) zum Nachweis von minimaler Resterkrankung (minimal residual disease, MRD) etabliert und für umfassende begleitende Untersuchungen eingesetzt. Für das FCM-MRD Monitoring von BCP-ALL wird eine 8-Farb-Kombination von Antikörpern eingesetzt, die neben der MRD-Analyse eine detaillierte Beschreibung des Regenerationsstatus vom Knochenmark des Patienten erlaubt.
Unsere Analyse der MRD-Proben einzelner Patienten zeigte eine starke interindividuelle Variabilität hin-sichtlich der Intensität und Reifungsstufe der hämatopoetischen Regenera-tion. Von besonderem Interesse, in einigen Fällen konnten wir neben CD19-positiven B-Zell-Vorläuferzellen auch die oben beschriebene CD34+CD38-CD19- Zellfraktion identifizieren, die eine Positivität für CD 22 (B-Zell Marker) und eine aberrant hohe Expression vom CD10-Antiogen (ein Merkmal von BCP-ALL) aufwiesen. Damit handelt es sich um eine sehr kleine Subpopulation der Zellen im Knochenmark (ca.10 - 100 Zellen aus insgesamt 1.000.000 untersuchten Zellen pro Knochenmarkprobe), die sowohl immunphänotypische Eigen-schaften von LSCs als auch von sehr frühen B-Vorläuferzellen aufweisen.
Im beantragten Vorhaben soll die Frage beantwortetet werden, ob eine Korrelation zwischen der Anwesen-heit bzw. Frequenz dieser LSC-ähnlichen Zellen bei Diagnosestellung und dem klinischen Verlauf der Er-krankung vorliegt. Darüber hinaus sollen diese Arbeiten eine Grundlage bilden für umfassende Unter-suchungen der LSC-ähnlichen Zellen bezüglich ihrer molekular-biologischen Eigenschaften sowie ihres Potentials als therapeutisches Angriffsziel in ALL des Kindesalters.
nach oben zur Projektseite home14/9
Molekularbiologische Detektion von Aspergillus fumigatus und neuen CYP51A- assoziierten Azol-Reistenz-vermittelnden Mutationen in brochoalveolären Lavage- (BAL) und Blutproben von Patienten mit fortgeschrittenem MDS
Dr. sc. hu.
Birgit Spiess, Molekularbiologin / Prof. Dr. med. Dieter Buchheidt,
Oberarzt,
III. Med. Klinik Universitätsmedizin Mannheim - Wissenschaftliches
Labor, Pettenkoferstr. 22,
68169 Mannheim
Patienten mit langdauernden
Neutropenie-Phasen, also auch Patienten mit fortgeschrittenem
myelodysplastischen Syndrom (MDS), haben ein sehr hohes Risiko, an
systemischen Pilzin-fektionen, in erster Linie bedingt durch
Aspergillus fumigatus, zu erkranken. Die infektionsbe-dingte
Sterblichkeit dieser Infektionskomplikation ist, trotz neuer Diagnostik-
und Therapie-möglichkeiten, immer noch hoch. Neben der Verbesserung der
frühzeitigen Diagnostik invasiver Pilzinfektionen ist es auch von
zunehmender klinischer Relevanz, Resistenz-mechanismen gegen
Antimykotika nachzuweisen, um die spezifische Therapie zu optimieren.
Ziel des geplanten Projektes ist die frühzeitige, molekularbiologisch-genotypisch basierte Detektion weiterer Mutationen im gesamten Cytochrom-51A-Gen (cyp51A) des fungalen humanpathogenen opportunistischen Erregers Aspergillus fumigatus, die Resistenzen gegen standmardmäßig eingesetzte Triazol.-Antomykotika bewirken. (Etwa zwanzig Aspergillus-Arten werden als humanpathogene und opportunistische Krankheitserreger eingestuft, wobei Aspergillus fumigatus die häufigste ist.)
Zur Detektion neuer und zur Optimierung der Detektion publizierter Mutationen im Aspergillus fumigatus cyp51A Gen sollen, zusätzlich von den von unserer Arbeitsgruppe bereits etablierten Assays, weitere sensitive und spezifische PCR-Assays (mit konsekutiver ‚nachfolgender‘ Sequenzanalyse) entwickelt werden. Die molekularbiologische Untersuchung soll in einem innovativen Ansatz –nicht kulturbasiert- direkt an geeignetem klinischen Material erfolgen.
Die sensitive und kulturunabhängige Detektion Resistenz-verursachender Mutationen könnte die Wahl eines effektiven Antimykotikums erleichtern und durch die Optimierung der antimykotischen Therapie zu einer Verbesserung der Prognose von Patienten mit lang-dauernder Netropenie bei fortgeschrittenem MDS führen.
Zielsetzung ist die methodische Entwicklung
weiterer sensitiver und spezifischer (nested) PCR-Assays mit
konsekutiver Seqenzanalyse zur Detektion neuer und publizierter
Mutationen im gesamten Aspergillus-fumigatus cyp51A Gen, die eine
Resistenz gegen
standardmäüig therapeutisch eingesetzte Triazol-Antimykotika bedingen,
die molekularbiologische Untersuchung erfolgt – nicht kulturbasiert-
direkt am klinischen Material.
nach oben
zur Projektseite
home
14/10
Assistenzsysteme zur Erhöhung der Sicherheit bei
myelodysplastische Syndrom und myeloproliferative Neoplasien
Ralf Bieber M. Sc., III. Med. Klinik
Universitätsmedizin Mannheim - Wissenschaftliches Labor, Pettenkoferstr.
22, 68169 Mannheim
(Definition aus Wikipedia)
Translationale Medizin ist die Schnittstelle zwischen präklinischer
Forschung und klinischer Entwicklung. Sie beschäftigt sich mit der
Übersetzung von z. B. in-vitro- Modellen oder Tiermodellen in die
Anwendung am Menschen.
Die heutige medizinische Diagnostik ist durch die vielen Erfolge der
Translationalen Medizin und der Trans-lationalen Bioinformatik zu einem
Feld geworden, in dem die schnelle und richtige Verarbeitung von Daten
einen immer größeren Stellenwert einnimmt. Ganze Krankheitsbilder (z. B.
die Chronische myeloische Leukämie) mussten durch die Klärung der
molekularen Ursachen in Diagnose und Therapie fundamental verändert
werden. Die Aufklärung der molekularen Ursachen ist ein wichtiger Aspekt
der Translationalen Medizin, wobei das Verständnis der produzierten
Daten und eine Hilfestellung beim Bewerten der Zusammenhänge für
medizinisches Fachpersonal nur noch mit erheblichem Aufwand möglich ist.
Es gilt Assistenz bzw. Expertensysteme zu erforschen und Möglichkeiten
zu entwickeln, die diese Unterstützung leisten können. Diese Systeme
müssen mit einer klar verständlichen und intuitiv zu bedienenden
Benutzeroberfläche ausgestattet sein, um schnell und effektiv zur
richtigen Diagnose zu verhelfen.
Stethoskop, Abtasten, Anamnese, Medikation,
Temperatur, Blutbild, Mikroskop, sind von je her, neben der Erfahrung
des Arztes, die fundamentalen Werkzeuge zur Diagnosefindung.
Das moderne Diagnoselabor ergänzt mit zytogenetischen und
molekularbiologischen Daten, mittels verschiedenster technologischer
Ansätze (z. B. Polmerase Kettenraktion PCR,
Massenspektrometrie, DNA-Chips Microarray,
Next-Generation-Sequenzierung NGS.).
Diese Analyseverfahren geben Aufschluss über die genaue Abfolge
bestimmter Desoxy-ribonukleinsäure- (DNS) und Ribonukleinsäure- ((RNA)
Sequenzen und Mutationen, welche als Änderung der Sequenzfolge definiert
sind und dadurch eine immer größere Wichtigkeit in der Diagnosefindung
erfahren.
Die stetige Veröffentlichung neuer krankheitsassoziierter Mutationen
führt zu einem genaueren Verständnis des Krankheitsverlaufs, einer
besser aufgelösten Prognose und einer zielgerichteten Therapie.
Die Zusammenhänge sind sehr komplex. Das
Myelodysplastische Syndrom und Myeloproliferative Neo-plasien, die im
Feld der Molekulardiagnostik als Modellerkrankungen gelten, benötigt zur
Diagnose verschiedene molekularbiologische Analysen. Zur
Verlaufskontrolle, die alle sechs Monate empfohlen wird, sind mehrere
verschiedene molekularbiologische Analysen empfohlen.
Der direkte Zusammenhang der Ergebnisse dieser Analysen wird im Verlauf
mit allen vorhergehenden Analysen bewertet und führt nur bei richtiger
Auswertung zu einer optimalen Therapieentscheidung, die nach aktuellen
Daten zu einer ‚normalen‘ Lebenszeit der Patienten führen kann.
Andere Erkrankungen des blutbildenden Systems sowie die Entstehung
solider Tumore und weitere onko-logische Erkrankungen beruhen oft aus
Wechselwirkungs-Netzwerken, mutierten Genen sowie der genauen
Mutationsfrequenz der Gene untereinander, die in direktem diagnostischen
und therapeutischen Zusammenhang stehen. Die Abhängigkeiten und
Beziehungen sind sowohl von zeitlicher Natur in Form spezifischer
Mutationsstammbäume, wie in direkter Abhängigkeit, durch Begünstigung
oder Deaktivierung ganzer genomischer Bereiche.
Das Labor der III. Medizinischen Klinik der
Universitätsmedizin Mannheim ist akkreditiert für „Medizinische
Laboratorien mit besonderen Anforderungen an Qualität und Kompetenz“.
Einen der Grundpfeiler des Akkreditierungsprozesses stellt der Betrieb
der datenverarbeitenden Infrastruktur dar, der ein höchstes Maß an
Datensicherheit gewährleistet (IT Infra-structure Library Version 3
ITILv3). Es ist eine Sammlung von Best-Practice Anweisungen und ein
anerkannter Leitfaden, mit dem die bestmög-liche Verfügbarkeit von
IT-Diensten erreicht werden soll.
Durch die softwaregestützte Verwaltung der Laborprozesse und Optimierung der Abläufe bei gleichzeitiger Verbesserung der Qualität, konnte eine Halbierung der Zeit vom Probeneingang bis zum fertigen Befund erreicht werden. Die durchschnittliche Bearbeitungszeit wurde u. a. durch die Nutzung des Informationssystems von 12 auf 6 Werktage verbessert.
Das Projekt verfolgt folgende Ziele:
- Erweitern des Informationssystems um „wissensbasierte“ Methoden als
Grundlage zum
„Lernen“ und „Anwenden“ für wissensbasierte Modelle,
- Ausbau der Infrastruktur zur Beschleunigung
der Auswertung, bzw. Ausgleich des
Wachstums der Datenmengen,
- Verbesserung der Lehre und Förderung von
Nachwuchswissenschaftlern im Studium der
Medizinischen Informatik und Bioinformatik.
s. a. Projekt 2013/7
nach oben
zur Projektseite
home
14/11
Die Rolle von Prominin-1/CD133 für die
Antwort auf Tyrosin-Kinase Inhibitoren bei der Philadelphia
Chromosome-positiven aktuten lymphatischen Leukämie
PD Dr. Martin
Ruthardt, Med. Klinik II/Hämatologie, Klinikum der Goethe Universität,
Theodoer Stern Kai 7, 60596 Frankfurt am Main
aus Wikipedia:
Genexpression, auch kurz Expression oder Exprimierung, bezeichnet in
weitem Sinn, wie die genetische Information – eines
Gens (Abschnitt der
DNA) – zum Ausdruck kommt und in
Erscheinung tritt, also wie der
Genotyp eines
Organismus oder einer
Zelle als
Phänotyp ausgeprägt wird. Im engeren
Sinn wird darunter die Biosynthese von Proteinen anhand der genetischen
Information mitsamt allen dafür nötigen vorangehenden Prozessen
verstanden.
Die Resistenz
gegenüber Tyrosinkinase-Inhibitoren (TKI) ist die größte klinische
Herausforderung bei der Philadelphia-Chromosome positiven (Ph+)
Leukämie.
Während die chronische Phase der chronischen myeloischen Leukämie (CML)
mit Tyrosin-kinase-Inhibitoren gut beherrschbar ist, stellt die
Resistenz bei fortgeschrittenen Ph+ Leukämien wie der CML-Blastenkrise
und der Ph+akuten lymphatischen Leukämie (Ph+ und ALL) ein großes
Problem dar.
Das Philadelphia Chromosome ist das zytogenetische Korrelat der t(9;22),
welches zur Expression des BCR/ABL Fusionsproteins führt. BCR/ABL
induziert über seine aberrante Kinase-Aktivität den leukämischen
Phänotyp. Diese Kinase-Aktivität wird effektiv durch TKIs, wie Imatinib,
Nilotinib oder Dasatinib, gehemmt.
Für die Resistenz gegenüber den TKIs sind meistens Punktmutationen
verantwortlich, die verhindern, dass die TKI an BCR/ABL binden. Ein
gewisser Anteil an resistenten Patienten weist jedoch keine
Punktmutationen auf, welche die Resistenz erklären könnten. Bei diesen
Patienten bleibt der Mechanismus der Resistenz häufig unbekannt.
Ausgehend von der
Ergebnissen, welche gezeigt hatten, das das Fehlen von Prominin-1
(PROM-1) zu einer beschleunigten Induktion des leukämischen Phänotyps
durch BCR/ABL in Mäusen führt, konnten wir zeigen, dass PROM-1 eine
wichtige Rolle für eine korrekte Differenzierung aus dem
Stammzellkompartiment heraus spielt. Dies hat zur Folge, dass der
Verlust von PROM-1 zu einem höheren Anteil an ALLs (bis zu 50 %)
gegenüber der sonst beinahe 100 % an CMLs bei BCR/ABL-induzierten
Leukämien der Maus führt. Außerdem führt der Verlust von PROM-1 zu einer
„paradoxen“ Antwort auf den Kinase Inhibitor Imatinib in
BCR/ABL-positiven hämopoetischen Stamm- und Vorläuferzellen (HSPC).
Imatinib steigert den Anteil an primitiven unreifen HSPCs in diesem
Kompartiment. Da dies auf eine Rolle von PROM-1 nicht nur für das
„lineage comittment“, sondern auch in der nicht Mutation-assoziierten
Resistenz gegenüber TKIs hindeutete, haben wir die Expression von
PROM-1/CD133 in Modellen der nicht Mutation-assoziierten Resistenz der
Ph+ ALL untersucht.
Tatsächlich konnten wir zeigen, dass bei humanen Modellen der Ph+ ALL
ein direktes Verhältnis zwischen PROM-1 Expression und Antwort auf TKI
besteht. Bei vollständig TKI-resistenten Zellen war die PROM-1
Expression vollständig unterdrückt.
Ziel dieses Projektes ist die Aufklärung der Rolle von PROM-1 für die Biologie der Ph+ Leukämie weiter zu vertiefen und die Zusammenhänge zwischen PROM-1 Expression und der Antwort auf TKI bei der Ph+ALL aufzuklären.
Diese Untersuchungen
werden einen Beitrag zum besseren Verständnis der Biologie der
Ph+ Leukämie leisten und möglicherweise Wege aufdecken, die es erlauben
könnten, in Zukunft die Behandlung besser zu steuern und sogar zu
personalisieren
nach oben
zur Projektseite
home
geförderte Projekte 2015
15/1
Analyse von Dysregulationen der
DNA-Doppelstrangbruch-Reparatur als molekulares Prinzip für synthetisch
letale Therapieansätze bei myeloischen Neoplasien
Dr. med. Henning Popp, Prof. Dr.
Alice Fabarius, Universitätsklinik Mannheim
Dysregulationen der DNA-Doppelstrangbruch (DSB)-Reparatur sind wichtige
Mechanismen genetischer Instabilität, die zur Generierung komplexer
genetischer Veränderungen bei myeloischen Neoplasien (MN) beitragen
können.
Untersuchungen der vergangenen Jahre zeigten, dass Defekte der
DNA-DSB-Reparatur in Tumorzellen genutzt werden können, um diese gezielt
zu schädigen, indem Inhibitoren
(Hemmstoffe)
gegen Mechanismen verwendet werden, die den Defekt kompensieren und bei
Inaktivierung eine „synthetische Letalität" verursachen.
Ziel des Projektes ist es die
DNA-DSB-Reparaturkapazität bei verschiedenen MN zu untersuchen und
daraus neue, gezielte und weniger toxische Therapieansätze zu
entwickeln. Zur Analyse der DSB-Reparaturkapazität werden
γH2AX-Immun-fluoreszenz-färbungen an Zelllinien von MN und aufbereiteten
Blut-/ Knochenmark-proben von Patienten mit MN durchgeführt, die zuvor
mit Cisplatin (Mittel zur
Hemmung des Zellwachstums bzw. der Zellteilung)
bzw. γ-Strahlen zur Induktion von DSB behandelt wurden.
Die sich bildenden γH2AX-Foci (Herde,
Schwerpunkte)
werden mit Voranschreiten der Reparatur wieder abgebaut und als
Biomarker zur Analyse der DSB-Reparatur-Kinetik im zeitlichen Verlauf
verwendet.
Bei Nachweis einer Alteration (Umstellung,
Veränderung) der
DSB-Reparaturkapazität werden gezielt genomweite
DNA-Methylierungsanalysen und Hochdurchsatz-Sequenzierungen zur Analyse
des genetischen Defekts eingesetzt, und die Ergebnisse der
DSB-Reparatur-Analysen werden auch mit dem molekularen Genotyp von MN
korreliert (beschreibt eine
Beziehung zwischen zwei oder mehreren Merkmalen, Ereignissen, Zuständen
oder Funktionen).
Aus den Untersuchungen können wichtige Rückschlüsse über das Ausmaß von
Dys-regulationen der DNA-DSB-Reparatur bei verschiedenen MN gezogen
werden und die Erkenntnisse genutzt werden, um neue, synthetisch letale
Therapieansätze mit verschiedenen niedermolekularen Inhibitoren
(PARP1/2-, APE1-, ATM- und CHK1-Inhibitoren) in vitro bei MN zu
untersuchen
nach oben
zur Projektseite
home
15/2
Inhibition
der onkogenen Funktion des USE Binding Protein 1 (FUBP1)
als Therapieansatz für CML- und AML-Leukämien
Prof. Dr. Martin
Zörnig, Georg-Speyer-Haus Frankfurt am Main
FUBP1 ist ein Transkriptionsregulator, der
die Expression verschiedener Targetgene
kontrolliert. So wird z.B. das potente Onkogen
c-myc als
direktes Targetgen durch FUBP1 aktiviert (gezielte Genmodifikation).
Unsere Arbeitsgruppe konnte FUBP1 in einem Screen für neue
anti-apoptotische Onkoproteine identifizieren und die anti-apoptotische
Funktion dieses Proteins bestätigen.
Aus Wikipedia: (Apoptose ist eine Form des
programmierten Zelltods. Es ist ein „Suizid-programm“ einzelner
biologischer
Zellen. Dieses kann von außen angeregt werden (etwa durch
Immunzellen) oder aufgrund von zellinternen Prozessen ausgelöst
werden (etwa nach starker Schädigung der
Erbinformation). Im Gegensatz zum anderen bedeutenden Mechanismus
des Zelltods, der
Nekrose, wird die Apoptose von der betreffenden Zelle selbst aktiv
durchgeführt und ist somit Teil des
Stoffwechsels der Zelle. Dadurch unterliegt diese Form des Zelltods
strenger Kontrolle und es wird gewährleistet, dass die betreffende Zelle
ohne Schädigung des
Nachbargewebes zugrunde geht.
(Onkoproteine sind Proteine die
von entarteten/ mutierten Proto-onko-genen produziert werden
(unkontrolliertes Wachstum) und letztendlich schädlich sind.
Es ist nicht verwunderlich, dass FUBP1
als c-myc-aktivierendes
und gleichzeitig antiapoptotisch wirkendes Molekül in verschiedenen
soliden Tumoren des Menschen überexprimiert wird, u.a. in Lebertumoren
wie dem Hepatozellulären Karzinom (HCC; unsere eigenen Daten).
Wir konnten in Zellkultur- und in
vivo-Transplantationsversuchen zeigen,dass FUBP1 für die Entstehung und
das Fortschreiten von HCCs eine wichtige Rolle spielt. FUBP1 ist für die
Tumorigenität (tumorigene
Eigenschaft von Zellen ) von
HCC-Zellen essentiell, weil es anti-apoptotische und
zellzyklus-inhibierende Gene reprimiert. So konnten wir u.a. zeigen,
dass das wichtige Zellzyklusinhibitorgen
p21 ein
direktes FUBP1-Zielgen darstellt.
FUBP1 bindet mit einer entsprechenden "Furche" an das einzelsträngige
FUSE-DNA
Element, um benachbarte Gene zu regulieren.
Wir haben
verschiedene Substanzbibliotheken durchsucht und kleine Moleküle
identifiziert, die die FUBP1-Bindungsfurche besetzen und so die
Regulation von FUBP1-Zielgenen verhindern.
Eines dieser Moleküle erreicht in Kombination mit Standard-Chemotherapie
eine hervorragende anti-Tumorwirkung in Zellkultur und im
Xenograft-Tumormodell.
Zur Zeit wird der Einsatz dieses Moleküls bei HCC-Patienten in Kombination mit Standard-Chemotherapie erprobt.
In weiteren Studien konnten wir mit Hilfe
zweier FUBP1-defizienter Mausmodelle
nachweisen, dass das Protein für das sogenannte "Self renewal"
hämatopoietischer Stammzellen (LT-HSCs) essentiell ist. Der Selbsterhalt
dieser Stammzellen geht ohne FUBP1 verloren, und der LT-HSC-Pool
verschwindet durch eine erhöhte Apoptoserate und verlängerte
Generationszeiten der FUBP1-defizienten Stammzellen. Die Mausembryonen
sterben daher bei Abwesenheit von FUBP1 an Anämie. Ein Manuskript mit
diesen Daten befindet sich aktuell bei der Zeitschrift
Cell Reports
in Revision.
Unsere Ergebnisse, denen zufolge FUBP1 für den Erhalt normaler hämatopoitischer Stammzellen wichtig ist und gleichzeitig als Onkoprotein wirkt, legen die Vermutung nahe, dass FUBP1 auch für den Erhalt Chemotherapie-resistenter Leukämie-initiierender Zellen (LICs; leukämische Stammzellen) essentiell ist. Interessanterweise konnten wir mit Hilfe verfügbarer Datenbanken feststellen, dass die Expression von FUBP1 in chronisch myeloischen Leukämien erhöht ist im Vergleich zu normalem Knochenmarksgewebe. Zudem wird FUBP1 in AML-induzierenden Zellen (AML LICs) höher exprimiert als in LIC-depletierten AML-Zellpopulationen.
In dem geplanten Versuchsvorhaben sollen in vivo-Modelle für AML und CML durch genetische Manipulation und anschliessende Transplantation von Knochenmarks-zellen etabliert werden. Dabei werden wir durch sogenannte knockdown-Technologie wahlweise FUBP1 herunterregulieren und die dabei resultierende Leukämie-Ent-wicklung vergleichen mit Zellen, in denen FUBP1 nicht herunterreguliert wird. Ein erstes Vorexperiment in einem sehr aggressiven AML-in vivo-Transplantationsmodell hat gezeigt, dass nach Herunterregulation von FUBP1 ein Drittel der mit Bcr-Abl1 transformierten Zellen transplantierten Empfängermäuse überlebt, während alle Tiere, die transformierte Zellen mit normalem FUBP1 Expressionsniveau erhalten hatten, bereits nach 28 Tagen an einer CML verstorben sind. Dieses Experiment unterstützt die Arbeitshypothese, derzufolge FUBP1 in leukämischen Stammzellen eine essentielle Funktion als Onkoprotein aufweist.
Folgende Ziele werden experimentell verfolgt:
1. Nachweis der
onkogenen Wirkung von FUBP1 in AML- und CML-induzierenden
Zellen (leukämischen Stammzellen)
2. Untersuchung
des molekularen Mechanismus der onkogenen Wirkung von FUBP1
in Leukämien (Identifizierung der Leukämie-relevanten
FUBP1-Zielgene)
3. Ergänzung der
Mausdaten durch funktionelle Zellkulturassays mit humanen AML-
und CML-Zelllinien sowie Expressionsstudien in
Patienten-Biopsien
4. Entwicklung von Therapieansätzen mit
viralen FUBP1
shRNA-Konstrukten (AAV
bzw.lentiviral) bzw. mit den von uns bereits
entwickelten FUBP1-Inhibitoren
Zusammenfassung:
Mit dem vorgestellten Versuchsvorhaben soll die onkogene Wirkung von FUBP1 in leukämischen (AML und CML) Stammzellen untersucht und zu therapeutischen Zwecken (möglicherweise in Kombination mit Standard-Chemotherapie) inhibiert werden.
nach oben zur Projektseite home
15/3
Analyse genetischer Veränderungen in Mesenchymalen Stromazellen von Patienten im Hämatoplastischen Systemerkrankungen, insbesondere mit Myelodysplastischem Syndrom (MDS) in einem neuen in vivo Modell
Prof. Dr. med. Wolf-Karsten Hofmann, III. Med. Klinik Hämatologie und Onkologie, Mannheim
Myelodysplastische Syndrome (MDS) sind eine Gruppe heterogener maligner Knochenmarkerkrankungen, die durch eine ineffektive dysplastische Hämatopoese mit peripheren Zytopenien, und einem stark erhöhten Risiko einer Transformation in eine sekundäre akute myeloische Leukämie (AML) gekennzeichnet sind. Die exakte Diagnosestellung und Einordnung in Klassifizierungssysteme erlauben eine Einstufung der hetero-genen Erkrankung nach Risiko, was wiederum Konsequenzen für die Art der Therapie hat. Diese kann von einer rein palliativen, unterstützenden Therapie über Wachstumsfaktoren, DNA-demethylierender Substan-zen, immunmodulatorischer Wirkstoffe wie Lenalidomid bis hin zu einer allogenen Stammzelltransplantation in kurativer Absicht reichen.
Bezüglich der Pathogenese wird eine schrittweise Anhäufung von genomischen Schäden wie chromoso-maler Aberrationen, DNA Punktmutationen und epigenetischer Veränderungen in den hämatopoetischen Stammzellen vermutet, die im Verlauf zum Auswachsen des MDS im Knochenmark führen. Trotz Identif-ikation zahlreicher solcher genomischer Schäden in jüngster Vergangenheit bleiben die dadurch vermittelten molekularen Mechanismen der MDS weitgehend unverstanden, da im Gegensatz zu anderen hämatolo-gischen Erkrankungen bisher kein geeignetes in vitro oder in vivo Modell zur Durchführung biologisch-funktioneller Analysen zur Verfügung stand. Die Etablierung von MDS Zellkulturen oder Xenograftmodellen scheiterte bisher an den scheinbar schlechten Wachstumseigenschaften von primären MDS Zellen, so dass bisher nur vereinzelt ein „Engraftment“ (Anwachsen eines Transplantats) von Hoch-Risiko MDS-Zellen in sogenannten immundefizienten (immungeschwächten) Mäusen erreicht werden konnte.
Das vorliegende Projekt bezieht sich auf die Hypothese, dass das MDS nicht eine isolierte Erkrankung der hämatopoetischen Stammzellen ist, sondern auch auf eine deregulierte Funktion der Knochenmarknische zurückzuführen ist. Um diesen Zusammenhang weiter untersuchen zu können, wurde weltweit erstmalig ein entsprechendes in vivo Modell etabliert. Die Arbeiten an diesem Modell sind extrem aufwendig und ziehen eine sehr große Anzahl an Zellkulturexperimenten nach sich.
Finanziert werden zwei CO2-Inkubatoren HERACELL 150 l (VWR), da für die langfristige und planbare Durchführung des Projektes eine umfangreiche Kultivierung von hämatopoetischen Stammzellen (kommen primär im Knochenmark vor und sind Ausgangspunkt für die Zellneubildung des Blutes) und von mesenchymalen Stromazellen (mesenchymale stromale Zellen sind multipotent und unterstützen Wachstum und Entwicklung der Vorläuferzellen des Blutes im Knochenmark) von jedem zu untersuchenden Patienten erforderlich ist. Die Kulturen dieser Zellen werden über mehrere Wochen durchgeführt, wodurch es zu einer starken Steigerung der Anzahl von Zellkulturen kommt, die in entsprechenden CO2-Inkubatoren gehalten werden müssen.
nach oben zur Projektseite home15/4
Charakterisierung von molekularen Kofaktoren bei der aktuten lymphatischen Leukämie (ALL) des Erwachsenenaleters mit Alteratin des MLL-Gens
PD Dr. med. Dr. rer. nat. Thomas Burmeister,
Facharzt für Innere Medizin mit Schwerpunkt
Hämatologie und internistische Onkologie, Laborleiter Tumorgenetik im
Labor Berlin – Charité
Vivantes Arbeitsgruppenleiter Charité, Medizinische Klinik für
Hämatologie, Onkologie und Tumorimmunologie, Hindenburgdamm 30. 12200
Berlin
Diese genetische Vielfalt hat auch klinische Konsequenzen. ALL-Patienten, die unterschiedlichen geneti-schen Gruppen angehören, zeigen einen sehr unterschiedlichen klinischen Verlauf. Manche lassen sich mit einer herkömmlichen intensiven Chemotherapie heilen ("Standardrisiko"-Patienten), bei anderen reicht diese nicht aus und nur ein vollständiger Ersatz der erkrankten Blutbildung durch die Blutbildung eines passenden Spenders ist erforderlich (allogene Transplantation), um eine Heilung erzielen zu können ("Hochrisiko"-Patienten).
Mit dem zunehmenden Wissen um die genetischen Grundlagen der ALL wird immer mehr klar, dass die bisherige Einteilung in Standardrisiko- und Hochrisiko-Patienten anhand der bekannten Risikofaktoren immer noch viel zu grob ist. Auch unter den StandardrisikoPatienten gibt es solche, die einen sehr ungünstigen Verlauf aufweisen und von den Hochrisiko-Patienten müssen wahrscheinlich nicht wirklich alle der risiko-reichen allogenen Transplantation zugeführt werden.
Die Erkenntnisse der Genetik haben zudem die Entwicklung von tumorspezifischen Medikamente möglich gemacht, die relativ gezielt auf Tumorzellen wirken und damit nicht so sehr wie herkömmliche Chemo-therapeutika (Zytostatika) auch das gesunde Gewebe schädigen.
Das vorliegende Projekt befasst sich mit einer
Untergruppe der akuten lymphatischen Leukämie im Er-wachsenenalter, und
zwar den Patienten mit Veränderungen des MLL-Gens auf Chromosom
11. Diese Patienten zählen als Hochrisiko-Patienten, zeigen aber auch
keinen einheitlichen Krankheitsverlauf. Bisher ist wenig über andere
begleitende genetische Veränderungen in dieser Patientengruppe bekannt.
Diese sollen im Rahmen des Projektes untersucht werden. Im engeren Sinne
sollen Veränderungen in den drei Genen NRAS, KRAS und
FLT3 analysiert werden. Es soll nach Mutationen, die die Funktion
dieser Gene beeinträchtigen, geforscht werden und klinisch untersucht
werden, welche Auswirkungen solche Mutationen auf den Verlauf der
Erkrankung haben.
nach oben zur Projektseite home
15/5
Genetische Alterationen in Mesenchymalen Stromazellen von AML Patienten
Prof.
Dr. med.
Claudia Baldus, Charité, Campus Benjamin Franklin, Hämatoligie,
Onkologie, Tumorimmunologie, Hindenburgdamm 30, 12203 Berlin
Die
molekulargenetische Charakterisierung von akuten Leukämien hat
wesentlich
zum Verständnis dieser sehr bösartigen Erkrankung des blutbildenden
Systems beigetragen.
In den letzten Jahren konnten so verschiedene molekulare Veränderungen
aufgedeckt werden, die helfen, das Risikoprofil der Leukämie besser
vorherzu- sagen und zudem die Grundlage für die Entwicklung von neuen
Therapieansätzen bilden. Das Ziel bleibt, die Heilungschancen von
Patienten mit akuter Leukämie weiter zu verbessern.
Diese bisherigen Analysen zur Identifizierung von molekularen
Veränderungen beschränken sich überwiegend auf die Untersuchungen der
Leukämiezellen. Es gibt jedoch Hinweise, dass zusätzlich auch
Veränderungen in den die Leukämie-zellen umgebenden Stromazellen im
Knochenmark vorkommen.
So können durch
genetische Veränderungen der Stromazellen im Knochenmark und einer damit
einhergehenden Veränderung des Knochenmarkmilieus aus gesunden
blutbildenden Vorläuferzellen Leukämiezellen entstehen. Die durch das
schützende Knochenmarkstroma vermittelte Chemotherapieresistenz ist von
wesentlicher klinischer Relevanz.
So können die unzureichenden Therapieerfolge unter anderem auf den
schützenden Einfluss des Knochenmarkstromas auf die Leukämiezellen
zugeschrieben werden.
Die Untersuchung des Knochenmarkstromas von Leukämiepatienten soll ein
Schwerpunkt des Forschungsprojektes sein. Durch genomweite Analysen
sollen genetische Veränderungen in Stromazellen von Patienten mit akuter
myeloischer Leukämie (AML) identifiziert werden.
In dem hier beantragten Projekt möchten wir nun nicht manipulierte, d.h.
frisch isolierte Knochenmarkstromazellen von Leukämie Patienten ebenso
wie von gesunden Probanden untersuchen.
15/6
Etablierung der Echtzeit-Deformationszytometrie als neuartiges diagnostisches Verfahren bei myledysplastischen Syndromen
Prof. Dr. Uwe Platzbecker, Med. Fakultät Carl Gustav Carus an der TU Dresden, Med. Klinik und Poliklinik I, Fetscherstr. 74, 01307 Dresden,
Prof. Dr. Jochen Guck, Biotechnisches Zentrum
BIOTEC, TU Dresden, Tatzberg 47/49, 01307 Dresden
Die Routinediagnostik der MDS schließt eine
zytomorphologische und histologische Beurteilung des Knochenmarks,
zytogenetische und gegebenfalls molekular-biologische und
durchflusszytometrische Untersuchungen ein.
Trotz dieser aufwendigen Verfahren sind die MDS häufig
Ausschlussdiagnosen und mit etablierten Verfahren anfänglich nicht zu
beweisen. Dies wird von vielen Patienten als belastend beschrieben und
kann zu einer Verzögerung einer zielgerichteten Behandlung führen. Daher
besteht ein großer Bedarf an diagnostischen Methoden, die eine
zeitnahe und sichere Diagnosestellung erlauben.
Die Echtzeit-Deformationszytometrie (Real-Time
Deformability Cytometry, RT-DC) ist eine in diesem Zusammenhang
bisher noch nicht verwendete Methode mit großem Potential. Sie erlaubt
die schnelle mechanische Charaktersierung von Zellen ohne vorherige
Markierungsschritte.
In ersten Vorversuchen konnten wir bereits zeigen, dass MDS-Stammzellen
über bestimmte Merkmale verfügen, die zu einer veränderten Zellmechanik
führen.
Diese Ergebnisse sollen in einer größeren Kohorte validiert werden und
gleichzeitig soll evaluiert werden, ob sich RT-DC zur MDS-Diagnostik
eignet.
15/7
Softwaregestütztes Erkennen u9nd Bewertung von Mutationsformationen aus traditionellem und High Trougput Sreening
Ralf Bieber M. Sc., Prof. Dr. med. Wolf-K. Hofmann, Universitätsmedizin Mannheim. III. Med. Universitäts-klinik, Hämatologie und Onkologie, Wissenschaftliches Labor, Pettenkofer Str. 22, D-68169 Mannheim
Das Labor der III. Medizinischen Klinik der Universitätsmedizin Mannheim
befasst sich mit der Erforschung und Erkennung der molekularen Ursachen
onkologischer Veränderungen des blutbildenden Systems. Die Ergebnisse
dieser Translationalen Forschung führen zu einer verbesserten
Diagnose und Therapiekontrolle der betroffenen Patienten.
Unter Verwendung moderner molekularbiologischer Verfahren werden
spezifische qualitative und quantitative Aussagen über das Vorkommen
krankheitsassoziierter Mutationen erstellt und als Befund dem
anfordernden Arzt mitgeteilt.
Aufgrund großer Fortschritte im Bereich der Analyseverfahren steigt die
Zahl der bekannten Mutationen einer Erkrankung in einem
exponentiellen Maß.
Die grundlegende Aufgabe des Klassifizierens und Zuordnens der Bedeutung
einer Mutation ist ein komplexer Prozess, der die Recherche in Literatur
und Internetdatenbanken erfordert. Des weiteren sind Informationen zur
Bestimmung der Auswirkungen einer Erkrankung in verschiedenen Quellen
oft widersprüchlich festgehalten.
Aufgrund des Maschinenoutputs von ca. 1000 möglichen Mutationen je
Analyse ist dieser Prozess ohne softwaregestützte Assistenzsysteme nicht
mehr handhabbar.
Dieses Projekt befasst sich mit Methoden zur automatisierten Erfassung
und Klassifizierung von öffentlich verfügbaren Daten sowie dem Erstellen
einer Referenzwissensbasis zum automatisierten Erkennen von Mutationen.
Ziel ist
1. „Lernen“ und „Anwenden“ für wissensbasierte Modelle (siehe Projekt 10/2014)1)
2. Erweiterung der Funktionalität um eine Datenbasis zur Erkennung genetischer Informationen und Klassifizierung von genomischen Mutationen
3. Integration und
Erweiterung von Assistenzsystemen mit dem Ziel der Unter-
stützung bei der Erstellung sowie Interpretation
molekularer Laborbefunde
Die Umsetzung erfolgt durch Anwendung des generierten Wissensmodells in Programmcode zur Unterstützung bestehender Datenbanken und Funktionen.
Finanziert werden zwei Laptop’s, ein Rechenknoten und zwei Jahresverträge für
wissenschaftliche Hilfskräfte, da der Betrieb durch die größere potentiell zu verarbeitende Datenmenge die Anforderungen an Ausbau, Wartbarkeit und Komplexität der Infrastruktur erfordert nach oben zur Projektseite home
15/8
Analyse genetischer Veränderungen in mesenchymelan Stromazellen von patienten mit myelodysplasischem Synadrom (MNDS) in einem neujen MDS in-vivo-Modell
Prof.
Dr. med. Wolf-K.
Hofmann, PD Dr.
med. Daniel Nowak, III. Med. Klinik Hämatologie und Onkologie,
Universitätsmedizin, Theodor-Kutzer-Ufer 1 – 3, 68167 Mannheim
Beim myelodysplastischen Syndrom (MDS) handelt
es sich um eine Modellerkrankung der Hämatopoese. Durch verschiedene
zellbiologische und molekulargenetische Methoden konnte gezeigt werden,
dass für die leukämische Transformation der hämatopoetischen Stammzelle
die Kumulation einer Vielzahl von endo-genen und exogenen Ereignissen
erforderlich ist. In einem kürzlich von der Arbeitsgruppe Nowak/Hofmann
in Kooperation mit dem Deutschen Krebs Forschungs Zentrum (DKFZ,
Arbeitsgruppe Trumpp) etablierten in-vivo-Modell für das MDS konnte
erstmals das Anwachsen typischer MDS-Hämatopoese erreicht werden.
Im vorliegenden Projekt sollen genetische Veränderungen in
hämatopoetischen als auch in mesenchymalen Stromazellen von
MDS-Patienten, welche in das in-vivo-Modell transplantiert worden sind,
untersucht werden.
Ziel der Arbeiten für dieses Projekt ist es, die (gestörte?) Interaktion
zwischen blutbildenden Zellen und Knochenmarkbindegewebe genau zu
analysieren. Damit könnten grundlegende neue Erkenntnisse über die
Pathophysiologie des MDS gewonnen werden.
15/9
Unterstützung des Deutschen Registers für Stammzelltransplantationen DRST
Das Deutsche Register für Stammzelltransplantationen (DRST), gegründet
am
03. 04.1998 in Frankfurt am Main, ist zuständig für die zentrale
Erfassung und standardisierte Auswertung aller in deutschen
Transplantationszentren nach dem 01.01.1998 durchgeführten
Transplantationen und liefert zeitnah wichtige Daten über durchgeführte
Transplantationen bei verschiedenen Indikationen.
Damit stehen den Transplantationszentren wichtige Referenzgrößen zur Beurteilung, wie auch zur Planung weiterer Aktivitäten und Studien zur Verfügung.
Darüber hinaus unterstützt das DRST die Durchführung von nationalen und internationalen wissenschaftlichen Studien aktiv.
Es arbeitet eng mit dem europäischen Zentrum
(European Group for Blood and Marrow Transplantation, Maastricht,
Niederlande) zusammen. Dort werden
europaweit alle Blutstammzelltransplantationen in einer Datenbank
gespeichert
geförderte Projekte 2016
Förderpreis 2016 für wissenschaftliche Arbeiten
16/1
Analyse genetischer Veränderungen in Mesenchymalen Stroma-zellen von Patienten im Hämatoplastischen Systemerkrankungen, insbesondere mit Myelodysplastischem Syndrom (MDS) in einem neuen in vivo Modell
Prof. Dr. med. Wolf-Karsten Hofmann, Dr. Daniel Nowak, III. Med. Klinik Hämatologie und Onkologie, Mannheim
Fortsetzungsförderung
Das
myelodysplastische Syndrom (MDS) ist eine klonale maligne Erkrankung der
Hämatopoese. Das Krankheitsbild ist gekennzeichnet durch eine periphere
Zytopenie, hier insbesondere führend eine Anämie. Die Erkrankung
betrifft vor allen Dingen Patienten in fortgeschrittenem Alter, das
mediane Alter bei Erstdiagnose beträgt ca. 70 Jahre.
Die Standardbehandlung des MDS besteht in der Transfusion von
Erythrozytenkonzen-traten. Eine Vielzahl von experimentellen Therapien
von Wachstumsfaktoren über demethylierende Substanzen bis hin zu
immunmodulatorischen Medikamenten kommen zur Anwendung, um das Ausmaß
der hämatopoetischen Insuffizienz zu verringern.
Die einzige kurative Therapieoption für Patienten mit MDS besteht
zurzeit in der allogenen Stammzelltrans-plantation.
Die Pathogenese des MDS ist bis heute nicht geklärt. Sieht man von den
wenigen Fällen sogenannter sekundärer MDS ab, welche durch die
langanhaltende Exposition des betroffenen Patienten zu benzol-haltigen
chemischen Stoffen bedingt sein können, kann in über 95 % der Fälle eine
Ursache der Erkrankung nicht festgestellt werden.
Ein großes Problem und ein die Prognose bestimmender Umstand ist, dass die frühe Form des myelo-dysplastischen Syndroms, die in der Regel nur durch eine Anämie, Bi- oder Trizytopenie gekennzeichnet ist, sich im Verlauf der Erkrankung verändern kann und es zur Entwicklung einer akuten myeloischen Leukämie kommt. Durchschnittlich 30-50% der Patienten erleiden eine solche Transformation innerhalb von 3 Jahren. Es handelt sich dann in der Regel um eine Hochrisiko-Leukämie, die therapeutischen Optionen sind bei den älteren Patienten für diese Erkrankung sehr eingeschränkt.
Durch verschiedene zellbiologische und
molekulargenetische Methoden konnte gezeigt werden, dass für die
leukämische Transformation der hämatopoetischen Stammzelle die
Kumulation einer Vielzahl von endo-genen und exogenen Ereignissen
erforderlich ist. In einem kürzlich von der Arbeitsgruppe Nowak/Hofmann
in Kooperation mit dem Deutschen Krebs Forschungs Zentrum (DKFZ,
Arbeitsgruppe Trumpp) etablierten in vivo-Modell für das MDS konnte
erstmals das Anwachsen typischer MDS-Hämatopoese erreicht werden.
Das Modell wurde in CellStem publiziert.
siehe:
https://www.dkfz.de/de/presse/pressemitteilungen/2014/dkfz-pm-14-15-Kranke-Blutstammzellen-programmieren-ihre-Umgebung-um.php
Myelodysplastic cells in patients re-program mesenchymal stromal cells
to establish a transplantable stem cell-niche disease unit. Cell Stem
Cell 2014, DOI: 10.1016/j.stem.2014.02.014
PDF
Im vorliegenden Projekt sollen
genetische Veränderungen in hämatopoetischen als auch in mesenchymalen
Stromazellen von MDS-Patienten, welche in das in vivo-Modell
transplantiert worden sind, untersucht werden.
Z
nach oben
zur Projektseite
home
16/2
Quantifizierung von chromosomalen Veränderungen durch STR-Markeranalyse
bei Patienten mit myelodysplastischem Syndrom
Prof. Dr. med.
Wolf-Karsten Hofmann,
Cand.-Med.Johann-Christoph
Jann, III. Med. Klinik Hämatologie und Onkologie, 68167 Mannheim,
Theodor-Kutzer-Ufer 1-3
Die Diagnostik des myelodysplastischen Syndroms (MDS) schließt inzwischen neben der Morphologie der Knochenmarkszellen als unabdingbaren Bestandteil die Chromosomenanalyse mit ein. Eine klassische Chromosomenanalyse kann bei dieser Erkrankung ausschließlich aus Knochenmarkzellen durchgeführt werden. Die Prozedur der Knochenmarkpunktion ist relativ aufwendig und mit einigen Belastungen für den Patienten verbunden. Dieses Verfahren eignet sich also nicht, regelmäßig in kurzen Abständen bei den Patienten angewandt zu werden.
Für ein anhaltendes Monitoring des Krankheitsverlaufes ist die Untersuchung von peripherem Blut bei Patienten mit myelodysplastischem Syndrom bisher nicht gut geeignet gewesen, da in den peripheren Blutzellen typische Veränderungen, die sonst in Knochenmarkzellen mittels zytogenetischer und mole-kularer Analyse nachgewiesen werden konnten, nicht zu detektieren sind. Insbesondere die Chromosomen-analyse aus Zellen des peripheren Blutes bei Patienten mit MDS gestaltet sich als schwierig.
Die Diagnose MDS stützt sich auf zytomorphologische Untersuchungen des Knochen- marks und des peripheren Blutes. Ein Differentialblutbild sowie die Begutachtung eines peripheren Blutausstriches können bereits zu Beginn Hinweise auf die Ätiologie der Zytopenie geben, z.B. über den Nachweis von Virozyten, Formveränderungen der Erythrozyten, Blasten oder Dysplasiezeichen in der Granulopoese.
Die Chromosomenanalyse ist ein obligater diagnostischer Baustein und beeinflusst maßgeblich die Therapieentscheidung. Oft wird die Diagnose MDS auch erst durch den Nachweis einer bestimmten zytogenetischen Aberration bei nur geringen Dysplasie-zeichen und grenzwertiger Zytopenie gestellt. Chromosomale Veränderungen finden sich bei etwa 50% der neu diagnostizierten MDS-Patienten. Im Gegensatz zur konventionellen Zytogenetik mittels G-Bänderung können mit Hilfe der FISH (Fluoreszenz-in-situ-Hybridisierung)-Analyse spezifische numerische und strukturelle Chromosomen-aberrationen nicht nur an Metaphase-Chromosomen, sondern auch direkt im Interphasekern nachgewiesen werden. Dies ist speziell dann von Bedeutung, wenn nicht ausreichend Metaphasen gewonnen werden können.
Die Molekulargenetik mit ausgewählten Markern (z.B. TET-2, JAK-2, ASXL-1, EZH-2, RUNX-1, TP53, SF3B1) ist unterstützend in der Diagnosestellung als auch in der prognostischen Abschätzung des Krankheitsverlaufs.
Das hier vorgestellte Projekt soll eine molekulare, PCR-basierte, Technik entwickeln, mit welcher der Nachweis von initialen, durch zytogenetische Analyse dargestellte, chromo-somalen Veränderungen zur Diagnostik und zur Verlaufsbeobachtung bei Patienten mit MDS möglich ist.
Projektbezogene Vorarbeiten:
Analyse von "molekularen
Uhren" zur Bestimmung des mitotischen Zellalters in MDS und gesunden
Zellen,
Klonalitätsanalyse von Zellen in alternder gesunder und MDS Hämatopoese.
nach oben
zur Projektseite
home
16/3
Rolle des Insulin-ähnlichen Wachstumsfaktor-Rezeptors (IGF-1R)
in der Pathogenese der akuten myeloischen Leukämie
Prof.
Dr. Zhixiong Li, Prof. Arnold Ganser, Klinik für Hämatologie,
Hämostaseologie, Onkologie
und Stammzelltransplantation, Medizinische Hochschule Hannover,
Carl-Neuberg-Sr. 1
30625 Hannover
Proteintyrosinkinasen (PTKs) spielen eine
wichtige Rolle bei der malignen Transformation von Zellen. Bei vielen
Krebsarten sind Rezeptor-PTK dereguliert, daher könnten sie ein
wichtiges Ziel für eine molekulare Tumortherapie sein.
Bei der akuten myeloischen Leukämie (AML) war der bisherige Einsatz von monotherapeutisch wirksamen Inhibitoren der Tyrosinkinasen (z. B. FLT3-ITD oder c-Kit-Mutation) nur mit mäßigem Erfolg verbunden. Die Suche nach viel versprechenden molekularen Targets ist deshalb ein hoch aktuelles Thema.
Der Insulin-ähnliche Wachstumsfaktor-Rezeptor 1 (IGF1R) ist wichtig für die Entwicklung, Proliferation und Differenzierung aller Zellen und stellt ein Signalmolekül im Bereich der Apoptose dar. In humanen akuten Leukämien ist bisher wenig untersucht, ob IGF1R an der Entstehung dieser Erkrankungen beteiligt ist. Insbesondere wurden bisher keine in-vivo-Modelle von IGF1R-getriebenen Leukämie berichtet.
In einer Vorarbeit wurde die Phosphorylierung von IGF1R in 23% Patienten mit AML beobachtet. Die Antragsteller möchten deshalb in vivo-Modelle für IGF1R-getriebenen Leukämie etablieren, und die molekularen Mechanismen verstehen, die zur Entwicklung dieser Leukämien führen.
Die beschriebenen Arbeiten sollen helfen, die molekulare Therapie von AML in der Zukunft zu verbessern. nach oben zur Projektseite home
16/4
Unterstützung des Deutschen Registers für Stammzelltransplantationen DRST
Das Deutsche Register für Stammzelltransplantationen (DRST), gegründet am 3.4.1998 in Frankfurt am Main, ist zuständig für die zentrale Erfassung und standardisierte Auswertung aller in deutschen Transplantationszentren nach dem 01.01.1998 durchgeführten Transplantationen und liefert zeitnah wichtige Daten über durchgeführte Transplantationen bei verschiedenen Indikationen.
Damit stehen den Transplantationszentren
wichtige Referenzgrößen zur Beurteilung,
wie auch zur Planung weiterer Aktivitäten und Studien zur Verfügung.
Darüber hinaus unterstützt das DRST die Durchführung von nationalen und
internationalen wissenschaftlichen Studien aktiv.
Es arbeitet eng mit dem europäischen Zentrum (European Group for Blood
and Marrow Transplantation, Maastricht, Niederlande) zusammen.
Dort werden europaweit alle Blutstammzelltransplantationen in einer
Datenbank gespeicher
16/5
Hochdosiertes Cyclophosphamid nach allogener Knochenmark-transplantation von einem HLA-identen Spender bei älteren Patienten mit AML oder Hochrisiko-MDS
PD Dr. med. Gesine Bug, Med. Klinik II, Hämatologie und Onkologie, Universitätsklinik
Frankfurt am Main
Die allogene
Stammzelltransplantation (SZT) ist für viele Patienten mit einer akuten
myeloischen Leukämie (AML) oder einem myelodysplastischen Syndrom (MDS)
die Therapie mit der höchsten Heilungschance. Andererseits kann eine SZT
zu potentiell lebensbedrohlichen oder die Lebensqualität erheblich
einschrän-kenden Komplikationen führen, die vor.allem bei älteren
Patienten mit Komorbiditäten eine solche Therapie nicht ratsam
erscheinen lässt.
Ein besonderes Problem stellt die Graft-versus-Host-Erkrankung (GvHD)
dar, bei der die Spenderimmun-zellen u. U . schwere
Entzündungsreaktionen in fast allen Organen des Patienten auslösen
können.Eine akute (bis ca. Tag + 100 nach SZT) oder chronische GvHD
(nach Tag + 100) erfordert einen hochdosierten Einsatz von Steroiden in
Kombination mit einer langwierigen Behandlung mit zusätzlichen
Immunsuppres-siva und kann mit schwerwiegenden Folgeerkrankungen
einhergehen wie z. B. Niereninsuffizienz, Diabetes mellitus,
Fettstoffwechselstörungen, Bluthochdruck, Gefäßerkrankungen mit
zerebralen Durchblutungs-störungen, Osteoporose, Schimmelpilz- und
virale Infektionen u.v.m. Außerdem wird durch eine dauerhafte
immunsuppressive Therapie die antileukämische Wirkung des
Spenderimmunsystems der Graft-versus-Leukämie (GvL) - Effekt
beeinträchtigt, so dass die Wahrscheinlichkeit eines Leukämierezidivs
zunimmt.
Ein neues, innovatives Prinzip der GvHD-Prophylaxe stellt die hochdosierte Gabe des lymphotoxischen Präparates Cyclophosphamid (Cy) dar, das an den Tagen+ 3 und +4 (oder +5) nach einer allogenen Knochenmarktransplantation (KMT) verabreicht wird.
In diesem engen Zeitfenster reagieren die alloreaktiven lmmunzellen des Spenders, die die GvHD auslösen, so empfindlich auf Cyclophosphamid, dass im Falle eines vollständig HLA-kompatiblen Spenders bei vielen Patienten keine zusätzliche Immunsuppression nötig ist. .
Generell eignet sich die GvHD-Prophylaxe mit posttransplantations (post-Tx)-Cy auch für ältere Patienten, wurde aber bisher nur in Kombination mit anderen lmmun-suppressiva bei der haploidenten Familienspender-KMT eingesetzt, bei der zwischen Spender und Empfänger mehrere HLA-Differenzen bestehen.
Eigene Vorarbeiten
In der SZT-Einheit der Medizinischen Klinik ll setzen wir post-Tx-Cy in
Kombination mit Standardimmun-suppression seit 2011 erfolgreich für
Patienten mit hämatologischen Hochrisikoerkrankungen zur GvHD-Prophylaxe
ein, die ein Transplantat von einem Fremdspender mit mehr als einer
HLA-Differenz erhalten müssen, weil trotz dringender SZT-lndikation
weder ein HLA-kompatibler noch ein haploidenter Spender verfügbar ist.
Wir haben bis Ende Oktober 2015 insgesamt 25 Patienten mit dieser
Strategie transplantiert Eine erste Auswertung der viel versprechenden
Resultate bei 15 Patienten mit akuten Leukämien wurde bei einem
aktuellen Kongress (3. lnternational Congress on Controversies in Stem
Cell and Cellular Therapies - COSTEM, Oktober 2015 in Berlin)
präsentiert.
Bei den Fremdspendertransplantationen traten trotz zweier
HLA-Differenzen weder eine schwere akute noch chronische GvHD bei guter
antileukämischer Kontrolle auf. Diese Ergebnisse bilden die Grundlage
für die hier vorgeschlagene Phase ll-Studie.
nach oben zur Projektseite home
16/6
Einfluss der klonalen Hämatopoese im Transplantat gesunder Stammzellspender während der allogenen Stammzelltransplantation bei akuten Leukämien / MDS
Priv.-Doz. Dr. med.
Frederik Damm,
Charité, Campus Virchow, Abteilung Hämatologie, Onkologie,
Tumorimmunologie, Augustenburger Platz 1, 13353 Berlin, Germany
Für viele hämatologische Neoplasien ist die allogene Blutstammzelltransplantation die einzig kurative Therapiemöglichkeit. Als Spender werden nach Möglichkeit HLA-idente Geschwister eingesetzt. Wurden früher zumeist jüngere Patienten transplantiert, so steigt der Anteil der >60-jährigen Empfänger stetig an. Mit dieser Entwicklung eng verknüpft ist ein stetig steigendes Alter der Geschwisterspender.
Als klonale Hämatopoese
bezeichnet man einen prämalignen Zustand, der durch das Vorkommen von
Leukämie-assoziierten Mutationen im Blut gesunder Individuen definiert
ist und mit einem erhöhten Risiko
für hämatologische Neoplasien und einer erhöhten allge-meinen
Sterblichkeit einhergeht. Die klonale Hämatopoese tritt altersabhängig
auf und kann in mindestens 10-15% der > 60-jährigen nachgewiesen werden.
Der Einfluss der klonalen Hämatopoese im Spender bzw. im
Blutstammzelltransplantat auf das Transplan-tationsergebnis ist bisher
nicht erforscht, ist jedoch angesichts der wachsenden Zahl an älteren
Spender-Empfänger Pärchen ein hochaktuelles Thema.
Es ist bisher
völlig unklar, ob klonale Hämatopoese im Transplantat entscheidende
Faktoren einer allogenen Stammzelltransplantation, wie z.B. die
Engraftment-Zeit, schwere Infektionskomplikationen, Transfusions-bedarf,
Entwicklung einer Spenderzellleukämie, oder Abstoßungsreaktionen,
beeinflusst.
Da viele Mutationen der klonalen Hämatopoese zu einem erhöhten
Selbsterneuerungs- und Proliferations-verhalten der Blutstammzellen
führen, wären auch für den Empfänger günstige Effekte, wie ein
verstärkter Spender-gegen-Leukämie (GvL)- Effekt, durchaus vorstellbar.
In dem hier vorgestellten Projekt soll die DNA gesunder älterer (> 60 Jahre) Spender auf das Vorkommen von klonaler Hämatopoese untersucht und der Einfluss auf das Transplantationsergebnis systematisch analysiert werden. Hierzu werden ca. 300 Spender-Blutproben mittels eines targeted resequencing Ansatzes von 60 Leukämie-assoziierten Genen analysiert. Im Falle einer vermuteten Mutation wird diese in einem zweiten, unabhängigen Testverfahren bestätigt. Anschließend werden wir die klonale Dynamik und Expan-sion des mutierten Klons im Empfänger in Verbindung mit vorliegenden Chimärismus Analysen untersuchen. Abschließend erfolgt der Abgleich mit den klinischen Daten des Transplantationsverlaufs.
In dem hier
dargestellten Projekt soll also erstmalig der Effekt von klonaler
Hämatopoese im allogenen Transplantations-Setting untersucht werden, ein
Thema, welches angesichts der wachsenden Anzahl älterer Spender
zunehmend an Relevanz gewinnt.
siehe Veröffentlichung
https://ascopubs.org/doi/full/10.1200/JCO.2018.79.2184
nach oben zur Projektseite home
16/7
Teilautomatisierte- und qualitätsgesicherte- Molekulardiagnostik mittels softwaregestützter Assistenzsysteme
Prof. Dr. med. Wolf-Karsten Hofmann, Ralf Bieber, M. Sc, III. Med. Klinik Hämatologie und Onkologie, Wissenschaftliches Labor, 68169 Mannheim, Pettenkofer Str. 22
Die bestehende Datenbasis stellt sich aus
molekularen Werten folgender Technologien zusammen:
- Polymerase Kettenreaktion (PCR)
- Massenspektrometrie
- DNA-Chips (Microarray)
- Next-Generation-Sequencing (NGS)
Jede Technologie verfügt über eine Spanne an Einsatzmöglichkeiten und liefert spezifische Datenformate sowie Datenmuster. Aufgrund des Assistenzsystems für die Befundung stehen zusätzlich Therapie und Verlaufsdaten von ca. 19.000 Patienten mit einem mittleren FollowUp von 6 Proben zur Verfügung.
Myelodysplastische Syndrome und
Myeloproliferative Neoplasien, welche im Feld der Molekulardiagnostik
als Modellerkrankungen gelten, benötigen zur Diagnose verschiedene
molekularbiologische Analysen.
Zur Verlaufskontrolle, welche alle 6 Monate empfohlen wird, sind mehrere
verschiedene molekularbiologische Analysen empfohlen. Der direkte
Zusammenhang der Ergebnisse dieser Analysen wird im Verlauf mit allen
vorhergehenden Analysen bewertet und führt nur bei richtiger Auswertung
zu einer optimalen Therapieentscheidung, die nach aktuellen Daten, zu
einer „normalen“ Lebenszeit der Patienten führen kann. Andere
Erkrankungen des blutbildenden Systems, solide Tumore und weitere
onkologische Erkrankungen bestehen oft aus Wechselwirkungs-Netzwerken
mutierter Gene. Diese stehen oft in direktem diagnostischem und
therapeutischem Zusammenhang. Die Zusammenhänge und Beziehungen sind
sowohl von zeitlicher Natur in Form spezifischer Mutationsstammbäume wie
in direkter Abhängigkeit durch Begünstigung oder Deaktivierung ganzer
genomischer Bereiche.
Das Labor der III. Medizinischen Klinik der Universitätsmedizin verfügt über ein Labor- Informationssystem und Management System (LIMS), welches bereits eine Vielzahl an Assistenzsystemen bereitstellt, um die Laborprozesse optimal zu unterstützen. Grundlage dieser Unterstützung ist der Betrieb einer datenverarbeitenden Infrastruktur nach IT Infrastructure Library Version 3 (ITIL v3), welches ein höchstes Maß an Datensicherheit gewährleistet. ITIL ist eine Sammlung von Best-Practice Anweisungen und ein anerkannter Leitfaden für das IT- Service Management, mit dem die bestmögliche Verfügbarkeit von IT-Diensten erreicht werden soll.
Mittels modernster Continuous Integration- und DevOps-Ansätze (aus Wikipedia: DevOps beschreibt einen Prozessverbesserungs-Ansatz aus den Bereichen der Softwareentwick-lung und Systemadministration) verfügt das Labor über eine auf die Prozesse optimierte Software, welche sich an die vielfältigen Fragestellungen anpassen kann. Parallel wird durch das Einhalten strenger Programmiermuster und getesteter Frameworks die Qualität der Programmierung sichergestellt. Die Qualität der Software wurde aufgrund eines GCP Audits in einem Gutachten bestätigt.
Ziel ist
1. Die Erweiterung des Informationssystems
um weitere Assistenzsysteme zur
Automatisierung der Qualitätsparameter
2. Erweitern der Funktionalität um die
Erkennung studienassoziierter Fragestellungen
und Automatisierung der Abfragen
3. Integration und Erweiterung neuer Frameworks
in die IT Landschaft zur Steigerung der
Effizienz
Finanziert werden die Erweiterung der Rechen-
und Speicherkapazität, zwei Laptop’s,
ein Jahresvertrag für eine wissenschaftliche Hilfskraft, sowie die
Teilnahme an einem
wissenschaftlichen Kongress.
16/8
Unterstützung der halbjährlichen wissenschaftlichen Treffen der Kooperativen Transplantationsstudiengruppe 2016/2017 in Frankfurt
PD. Dr. Hans Martin, Universitätsklinik Frankfurt am Main, Med. Klinik II, Hämatologie/
Onkologie
nach oben zur Projektseite home
geförderte Projekte 2017
17/1
Unterstützung einer internationalen wissenschaftlichen Veranstaltung "46th Annual Scientific Meeting of the International Society for Experimental Hematology" 2017 in Frankfurt am Main
Prof. Dr. Michael Rieger, Universitätsklinik Frankfurt am Main, Med. Klinik II, Hämatologie/Onkologie
17/2
Dysregulationen der DNA-Doppelstrangbruch-Reparatur für molekulares Prinzip synthetisch letale Therapieansätze beim myelodysplastischen Syndrom
Prof. Dr. med Wolf-Karsten Hofmann, Dr. med. Henning Popp, III. Med. Klinik Universitätsmedizin Mannheim, Hämatologie und Onkologie, Theodor-Kutzer-Ufer 1-3, 68167 Mannheim
Dysregulationen der DNA-Doppelstrangbruch (DSB)-Reparatur stellen wichtige
Mechanismen genetischer Instabilität dar, die zur Generierung komplexer genetischer Veränderungen beim Myelodysplastischen Syndrom (MDS) beitragen können.
Untersuchungen der vergangenen Jahre haben gezeigt, dass Defekte der DNA-DSB-Reparatur in Tumorzellen genutzt werden können, um diese gezielt zu schädigen, indem Inhibitoren gegen Mechanismen verwendet werden, die den Defekt kompensieren, wodurch eine „synthetische Letalität“ verursacht wird.
Ziel des Projektes ist es, die DNA-DSB-Reparaturkapazität beim MDS zu untersuchen und daraus neue, gezielte und weniger toxische Therapieansätze zu entwickeln.
Zur Analyse der DSB-Reparaturkapazität werden Immunfluoreszenzfärbungen an aufbereiteten Blut-/Knochenmarkproben von Patienten mit MDS durchgeführt, die zuvor mit Cisplatin ((ein Arzneistoff Zytostatikum zur Hemmung des Zellwachstums bzw. der Zellteilung)) bzw. y-Strahlen zur Induktion von DSB behandelt wurden. Die sich bildenden yH2AX-Foci (Proteine als Marker für DNA-Brüche) werden mit Voranschreiten der Reparatur wieder abgebaut und als Marker zur Analyse der DSB-Reparatur-Kinetik im zeitlichen Verlauf verwendet. Bei verminderter DSB-Reparaturkapazität werden gezielt genomweite DNA-Methylierungsanalysen und Hochdurchsatz-Sequenzierungen zur Analyse des genetischen Defekts eingesetzt. Die Ergebnisse sollen als Grundlage genutzt werden, um neue, synthetisch letale Therapieansätze mit verschiedenen niedermolekularen Inhibitoren in vitro an Zelllinien und an CD34+ mononukleären Zellen von Patienten mit MDS zu untersuchen.
Ergebnis der Untersuchungen:
Analyse von Dysregulationen der DNA-Doppelstrangbruch-Reparatur als molekulares Prinzip für synthetisch letale Therapieansätze bei myeloischen Neoplasien
In dem Projekt wurden DNA-Doppelstrangbrüche
(DSB) und DSB-Reparaturmechanismen durch Immun-fluoreszenzmikroskopie
von γH2AX und 53BP1 in CD34+ Zellen von gesunden Individuen, von
Patienten mit myelodysplastischen Syndromen (MDS) und in Blasten von
Patienten mit akuten myeloischen Leukämien (AML) analysiert. Es zeigte
sich eine Zunahme der γH2AX Foci über das Spektrum von MDS zu AML sowie
eine Kolokalisierung der γH2AX und 53BP1 Foci.
Die Untersuchungsergebnisse unterstützen ein Modell der Karzinogenese
mit stufenweiser Anhäufung von DNA-Schäden und legen eine insuffiziente
Reparatur von DSB durch die nicht-homologe Rekombination nahe.
Weiterhin waren bestimmte Muster von γH2AX Foci in den Zellkernen
nachweisbar. Die γH2AX Foci waren im Zellkern diffus verteilt, in
Gruppen liegend oder randständig zur Zellkernmembran angeordnet. Die
Muster beruhen mutmaßlich auf einer nicht zufälligen, räumlichen
Genomorganisation bei MDS und AML.
Schließlich wurde die DNA-Schadensantwort in Blasten von AML analysiert.
Die DNA-Schadensantwort wird durch DNA-Schädigung aktiviert und wirkt
als „Anti-Krebs-Barriere“ beispielsweise durch Regulation des
Zellzyklus, der Transkription von Genen, der DNA-Reparatur und der
Apoptose. Es war eine Störung der DNA-Schadensantwort in Blasten von AML
mit einer reduzierten Aktivierung von Zellzykluskontrollpunkten und der
Apoptose nachweisbar.
Die Ergebnisse können genutzt werden, um neue gezielte therapeutische Ansätze durch synthetische Letalität bei MDS und AML zu untersuchen.
nach oben zur Projektseite home
17/3
Identifizierung von Onkogenen und Stammzellgenen durch Insertions-mutagenese
Prof. Dr. med. Zhixiong Li, Professor Dr.med. Arnold Ganser, Medizinische Hochschule Hannover,
Carl-Neuberg-Str.1, 30625 Hannover
Die Behandlung
der akuten myeloischen Leukämie (AML) von Erwachsenen ist
unbefriedigend. Die Identifizierung neuer Onkogene ist ein wichtiger
Schritt, den molekuraren Mechanismus für Leukämogenese zu verstehen und
effientere Therapien zu entwickeln.
Durch die Verwendung der Hochdurchsatz-Sequenzierung wurden immer mehr
genetische Mutationen bei Patienten mit Krebs entdeckt, was das
Verständnis für die Pathogenese der Leukämie wesentlich verbessert hat.
Allerdings dürfte die Liste der Gene, die als signifikant mutiert
identifiziert wurden, wahrcheinlich falsche Positives enthalten. Darüber
hinaus kann ein bedeutender Anteil der Onkogene durch Amplifikation
(Überexpression) und Aktivierung durch autokrine oder parakrine
Signalisierung induziert werden, welche nicht durch
Hochdurchsatz-Sequenzierung nachgewiesen werden können.
Wir haben die
sogenannte ”insertional dominande database” (IDDb) generiert, die
derzeit > 459 RVIS retrovirale Vektor-Inserionsstellen enthält und nach
einer langen Beobachtungszeit mit der klonalen Dominanz von normalen
oder leukämischen murinen hämatopoetischen (blutbildende) Zellen
isoliert.
In diesem Projekt möchten wir zwei Kanidaten Gene (PDGFRß und HOXB5) aus
unserer Datenbank untersuchen. Die Expression und/oder Aktivierung
dieser Gene in leukämischen Zellen wurde durch uns und andere gezeigt.
Die Bedeutung der beiden Gene in humanen akuten Leukämien ist allerdings
bisher wenig untersucht. Die Arbeiten sollen zu einem verbesserten
Verständnis der Pathophysiologie und neuen therapeutischen Konzepten bei
Leukämien führen.
17/4
Molekulargenetische Charakterisierung der akuten myeloischen Leukämie mit Translokation (8;21)
Priv.-Doz.
Dr. med. Frederik Damm, Charité, Campus Virchow, Abteilung Hämatologie,
Onkologie, Tumorimmunologie, Augustenburger Platz 1, 13353 Berlin,
Germany
Die akute
myeloische Leukämie (AML) ist eine bösartige Erkrankung des
blutbildenden Systems, die unbehandelt rasch zum Tode führt. Die
Translokation (8;21)(q22;q22) [kurz: t(8;21)] gehört mit ca. 10% zu den
häufigsten strukturellen Chromosomenaberration der AML und betrifft wie
die Inversion 16 eine Unter-einheit des Core-binding-factor-Komplexes,
was die Core-binding-factor-Leukämie (CBF-AML) definiert.
Die CBF-Leukämien zeichnen sich durch einen relativ günstigen klinischen
Verlauf bei Erstdiagnose aus – allerdings kommt es bei ca. 30% der
Patienten zu einem Rezidiv der Erkrankung, das meist eine hohe Resistenz
gegenüber Chemotherapie besitzt.
Eine Vielzahl genetischer Veränderungen und Mutationen, die zusätzlich zu der Translokation in den Leukämie-Zellen auftreten, wurden in den letzten Jahren identifiziert und bestimmen den klinischen Verlauf entscheidend mit. Während manche von Ihnen - z.B. Mutationen in den Genen KIT oder FLT3 - hinsichtlich ihrer prognostischen Bedeutung bereits detailliert untersucht worden sind, bleibt die Bedeutung zahlreicher Gen-Aberrationen noch unzureichend erforscht.
In dem
vorliegenden Projekt soll daher eine umfassende molekulargenetische
Analyse an der bislang größten Kohorte von 400 t(8;21) AML-Patienten
erfolgen. Dazu werden erworbene Mutationen in 60 Leukämie-relevanten
Genen mittels Tiefensequenzierung untersucht. Im Anschluss können genaue
Aussagen über Überlappung bzw. Exklusivität einzelner Mutationen erhoben
werden. Ferner können die klonale Zusammensetzung der Leukämie anhand
der Mutationslast einzelner Aberrationen für jeden Patienten ermittelt
und Rückschlüsse auf die genetische Leukämie-Entstehung erhoben werden.
Somit soll das Projekt einen wichtigen Beitrag auf dem Weg zu einem
personalisierten Therapieansatz der AML beitragen.
siehe Veröffentlichung
https://ashpublications.org/blood/article-lookup/doi/10.1182/blood-2018-05-852822
nach oben zur Projektseite home
17/5
Aufbau eines Registers zur Dokumentation der akuten GvHD im Rahmen des GvHD Registers der Deutsch-Österreichisch-Schweizer-Arbeitsgruppe GvHD
PD Dr.
Stefan A. Klein, III. Medizinische Klinik, Universitätsmedizin Mannheim,
Theodor-Kutzer-Ufer 1-3, 68167 Mannheim
Die akute Graft versus Host Disease (aGvHD)
ist die häufigste und schwer-wiegendste Komplikation nach allogenen
Stammzelltransplantationen. Je nach Spendertyp und Art der
Konditionierung erkranken zwischen 10 und 45% der allogen
transplantierten Patienten an einer aGvHD Grad III/IV. Insbesondere bei
Patienten mit gastrointestinaler Beteiligung ist der Verlauf der aGvHD
ungünstig und die Sterb-lichkeit hoch.
Für die Diagnostik und Therapie der akuten GvHD
existieren bislang keine durch prospektive Studien abgesicherten
Standards. Die meisten Daten zur Therapie der akuten GvHD entstammen
entweder retrospektiven Analysen, sind unizentrisch oder wurden vor mehr
als 20 Jahren erhoben und sind somit nicht auf die heutigen
Transplantations-modalitäten anwendbar. Das Fehlen einer fundierten
Datengrundlage ist einer der Hauptgründe für die Heterogenität im
Management der akuten GvHD. Jedes Transplantations-zentrum benutzt
eigene Standards. Es existieren erhebliche Unterschiede im Vorgehen
zwischen den Zentren
Um die Grundlage für ein einheitliches Vorgehen sowie für prospektive
Studien zur Therapie der aGvHD zu legen, ist es das Ziel dieses
Vorhabens ein Register zur Erfassung von Patienten mit akuter GvHD
aufzu-bauen.
Dazu wurde unter dem Dach der
Deutsch-Österreichisch-Schweizer-Arbeitsgruppe GVHD in den vergangen-en
zwei Jahren ein Register zur Erfassung der chronischen GvHD aufgebaut.
Hierzu haben die Transplan-tationszentren in Basel, Berlin-Buch,
Hamburg-UKE, Graz, Mainz, Mannheim, Regensburg, Wien und Wiesbaden eine
gemeinsame Registerordnung entwickelt und in Kraft gesetzt.
Es wurde ein webbasiertes eCRF für das Register entwickelt und durch die
Firma Evaluation Software Development, Rum bei Innsbruck, programmiert.
Die Kosten hierfür wurden von der Carsten Bender-Leukämie-Stiftung
aufgebracht. Darüber hinaus wurde das GvHD-Register mit dem Register der
EBMT (European Society for Blood and Marrow Transplantation) verzahnt.
So können die MedA-Datensätze aus dem EBMT-Register in das GvHD-Register
übernommen werden.
Aktuell wird das GvHD Register bereits aktiv von den Transplantationszentren in Basel, Graz, Jena, Mannheim, Nürnberg und Regensburg mit Daten gefüllt.
Das bereits bestehende Register zur Erfassung der chronischen GvHD soll um ein Modul zur Dokumentation der akuten GvHD erweitert werden. Ein verknüpftes Register für die akute und chronische GvHD ermöglicht die gemeinsame Nutzung der Stammdaten und der MedA-Daten der EBMT. Darüber hinaus können in einem gemeinsamen Register die nicht seltenen Spezialfälle der „late onset acute.“ der „progressive onset chronic GvHD“ und des „overlap syndrome“ zwischen akuter und chronischer GvHD gemeinsam erfasst werden.
Insbesondere soll das Modul zur akuten GvHD zusätzlich zu den MedA-Daten die folgenden Kategorien abbilden: Klinisches Bild und Grading der aGvHD bei Symptombeginn, bei maximaler Ausprägung im weiteren Verlauf und bei jedem Rezidiv der GvHD; Diagnostik; Histologie; Erfassung der Erstlinientherapie und jeder weiteren Therapielinie; Ansprechen auf die Erstlinientherapie und auf jede weitere Therapie; Outcome; Infektionen; Todesursache; Verlauf der Grunder-krankung sowie Laborbefunde und Biomarker.
Definitionen aus u. a.:
https://www.onkopedia.com/de/onkopedia/guidelinse/graft-versus-host-erkrankung
Die akute Graft-versus-host Erkrankung ist eine systemische, entzündliche Erkrankung, die nach allogener Blutstammzelltransplantation oder Knochenmarktransplantation auftritt und zur Schädigung insbesondere von Darm, Haut und Leber führt. Die aktuell gültige Definition des National Institute of Health unterscheidet
- klassische akute GvHD bis Tag 100
- „late-onset“ akute GvHD (nach 100 Tagen)
- persistierende und rekurrierende akute GvHD.
- chronische GvHD (ohne Zeitlimit)
- Overlap-Syndrom mit Zeichen der akuten chronischen GvHD
Die Programmierung durch die Firma Evaluation
Software Development, Rum, Österreich, wird durch die Alfred und
Angelika Gutermuth-Stiftung mit dem obigen Betrag ermöglicht.
Die Entwicklung und Programmierung des aGvHD-Moduls ist für das 2. und
3. Quartal 2017 geplant. Ab dem 4. Quartal 2017 soll das Register in
Betrieb gehen.
nach oben zur Projektseite home
17/6
Identifizierung molekularer Ziele des leukämischen Fusionsproteins RUNX1-ETO
Dr. rer. nat. Jörn Lausen, Forschungsgruppenleiter, Klinikum der Joh. Wolfg. Goethe-Universität,
Institut für Transfusionsmedizin und Immunhämatologie, Sandhofstr. 1, 60528 Frankfurt a. M
Die Hämatopoese (Blutbildung)
wird durch ein Netzwerk zelltypspezifischer Trans-kriptionsfaktoren
(Verlagerung von Chromosomenabschnitten t) gesteuert, welche
die Aktivität von Genen in der Zelle regulieren.
Funktionale Veränderungen dieser wichtigen Steuerungsmoleküle durch
Mutationen und chromosomale Translokationen sind mit Leukämieentstehung
assoziiert. Besonders häufig kommen in Leukämien die Veränderungen des
RUNX1-Gens vor.
RUNX1 ist ein Schlüsselregulator der
Hämatopoese. Er ist wichtig für die Entstehung von Stammzellen und die
Differenzierung und Reifung von Blutzellen.
Die chromosomale Translokation t(8;21) führt zur Expression
(Ausbildung des Phänotyps) des leukämie-assoziierten Fusionsproteins
RUNX1-ETO.
Dieses wird in ca. 40 % der AML (akute myeloide Leukämie)-Fälle des
M2-Typs bei Patienten gefunden. Obwohl RUNX1-ETO-abhängige Leukämien
intensiv erforscht werden, bleibt der genaue Mechanismus, wie RUNX1-ETO
zur Leukämieentstehung beiträgt, bislang ungeklärt.
Es wird angenommen, dass RUNX1-ETO an RUNX1-Zielgene bindet und dadurch
mit der normalen RUNX1-Funktion interferiert. In den von RUNX1-ETO
gebundenen DNA- Bereichen finden sich häufig so genannte E-Boxen. Diese
werden von Transkriptions-faktoren der
helix-loop-helix
Familie gebunden, wie z. B. TAL1, welches in der Zelle eine große Anzahl
von Gehen steuert.
TAL1 ist ein wichtiger hämatopoetischer Schlüsselregulator, notwendig
für die Bildung hämatopoetischer Stammzellen und die Entwicklung
verschiedener Blutlinien. TAL1 ist ein Onkogen (Krebs-Gen) bei
Leukämien, eine Fehlexpression trägt zur Leukämieent-stehung bei.
Da RUNX1-ETO-Komplexe auf potenziellen TAL1-Bindestellen in regulativen
Genelementen gefunden wurden, interferiert RUNX1-ETO möglicherweise mit
der TAL1 abhängigen Genexpression. Es liegt daher nahe, dass RUNX1-ETO
auf den betroffenen Genen mit E-Box bindenden Proteinen interagiert.
In den Vorarbeiten wurde gezeigt, dass RUNX1-ETO und TAL1 funktionell
miteinander verbunden sind.
In diesem Projekt soll untersucht werden, ob RUNX1-ETO in einem
Proteinkomplex auf regulativen Elementen von TAL1-Zielgene vorhanden ist
und dadurch das regulative transkriptionelle Netzwerk von TAL1 in der
Zelle stört. Dies würde zur Fehlregulation der Expression dieser Gene
und damit zur Leukämieentstehung beitragen. Das Aufdecken neuer
molekularer Ziele und bisher unbekannter Mechanismen der RUNX1-ETO
Wirkung bei AML könnte zur Entwicklung neuer Therapieansätze eingesetzt
werden.
17/7
Vorbereitung eines präklinikischen Modells zur Evaluation und Ent-wicklung neuer zielgerichteter Therapiestrategien bei Patienten mit myelodysplastischem Syndrom (MDS)
Prof. Dr. med Wolf-Karsten Hofmann,
Prof. Dr. med. Daniel Nowak, III. Med. Klinik Universitätsmedizin
Mannheim, Hämatologie und Onkologie, Theodor-Kutzer-Ufer 1-3, 68167
Mannheim
Die Erkrankung tritt mit einer Inzidenz von 3-10/100 000 pro Jahr auf und betrifft vorwiegend ältere Patienten mit einem starken Anstieg der Erkrankungsfälle auf >60/100 000 pro Jahr bei über Sechzigjährigen. Im Rahmen der demographischen Entwicklung gewinnt die Erkrankung daher immer mehr an Bedeutung. Klinisch präsentieren sich die Patienten mit Leistungsabfall, erhöhter Infektanfälligkeit und Blutungen, die durch die Zytopenien aller drei Zellreihen im Blut hervorgerufen werden.
In Vorarbeiten zu den hier beschriebenen Forschungsvorhaben ist es uns in einem Kooperationsprojekt unseres Forschungslabors mit der Abteilung Stammzellen und Krebs (Direktor, Prof. Andreas Trumpp) des DKFZ Heidelberg erstmals gelungen, ein robustes MDS Xenograftmodell mit MDS „low risk“ Entitäten zu etablieren. Die Ergebnisse der Arbeit konnten im April 2014 im Fachjournal „Cell Stem Cell“ publiziert werden.
Die Ergebnisse unserer bisherigen Forschungen liefern erstmals starke Indizien für eine pathogenetisch relevante Funktionseinheit zwischen hämatopoetischen Stammzellen und Knochenmarknischenzellen im MDS in einem in vivo Modell. Dieser Pathomechanismus könnte zum einen auch für andere hämatologische Neoplasien von Bedeutung sein. Zum anderen könnte die weitere Untersuchung des Modells ein Design neuer Therapiestrategien induzieren, die darauf abzielen, diese gestörte Nische in MDS zu manipulieren und fehlgeleitete Signalwege zu korrigieren. Davon abgesehen bietet die nun vorhandene Möglichkeit eines robust funktionierenden MDS Xenograft Modells neue Möglichkeiten für eine personalisierte Medizin für MDS Patienten. Während der Etablierung von Xenografts aus ihrem Knochenmark sind die Patienten noch am Leben. Es steht nun eine Plattform zur Verfügung um neue Substanzen und Zielgene für die Therapie von MDS zu testen.
Im hier beantragten Projekt soll nun dieses
Modell in konsequenter Fortsetzung der bisherigen Arbeiten eine
Plattform bieten, um neue Substanzen, deren Effektivität und
Zielstrukturen für die Therapie von MDS-Patienten zu testen und
ausgewählte Kandidaten für den klinischen Einsatz vorzubereiten.
Veröffentlichungen:
https://pubmed.ncbi.nlm.nih.gov/33054116/
https://pubmed.ncbi.nlm.nih.gov/31132434/
nach oben zur Projektseite home
17/8
Diagnostische Analyse von PDGFRB-Fusionsgenen bei malignen hämatologischen Erkrankungen
PD Dr.med. Dr. rer. nat. Thomas Burmeister, Charité CVK, Med. Klinik für Hämatologie,Onkologie und Tumorimmunologie, Augustenburger Platz 1, 13353 Berlin
Tyrosinkinasen (TK) sind Enzyme, die bei vielen Krebserkrankungen eine zentrale Rolle spielen. Unter normalen Umständen ist die Aktivität dieser Tyrosinkinase-Enyzme in menschlichen Zellen genau kon-trolliert und reguliert. Bei Krebserkrankungen findet man jedoch häufig eine unkontrollierte und stark erhöhte Aktivität einzelner Tyrosinkinasen in den Tumorzellen, die zum Beispiel dazu führt, dass die betreffenden Zellen unkontrolliert wachsen, sich unkontrolliert teilen und ihre normale Funktion nicht mehr erfüllen. Häufig wird dieses Außer-Kontrolle-Geraten der entsprechenden TK durch eine Chromosomentranslokation verur-sacht, bei der das betreffende Gen für die Tyrosinkinase mit einem anderen Gen fusioniert wird. Dadurch entsteht ein neuartiges Fusionsgen, das als Onkogen („Krebsgen“) wirkt und zu einem Protein führt, das die oben beschriebene gesteigerte und unkontrollierte Enzym-Aktivität aufweist.
Im Rahmen des vorliegenden Projektes soll die Tyrosinkinase PDGFRB untersucht werden. PDGFRB ist ein Rezeptor (Zelloberflächenprotein) für den Wachstumsfaktor aus Thrombozyten (Blutplättchen). PDGFRB spielt bei vielen bösartigen Bluterkrankungen und auch bei einigen Organtumoren eine wichtige Rolle. Zugleich ist dieses Enyzm auch von therapeutischem Interesse: es kann sehr effizient durch sogenannte Tyrosinkinase-Inhibitoren (TKI) gehemmt werden. Diese TKI werden als Tablette eingenommen und haben nicht das klassische, schwere Nebenwirkungsspektrum von Zytostatika, sondern sind in der Regel deutlich nebenwirkungsärmer, aber dabei trotzdem häufig wesentlich wirksamer als die letzteren.
Im Rahmen des Projektes soll die Genetik der PDGFRB-Fehlsteuerung in verschiedenen bösartigen Erkrankungen des blutbildenden Systems genauer erforscht werden. Ein besseres Verständnis dieser Veränderungen und bessere Möglichkeiten zum Nachweis dieser PDGFRB-Veränderungen bei Tumorerkrankungen würden die Behandlung der betreffenden Patienten erleichtern und verbessern.
nach oben zur Projektseite home
17/9
Entwicklung einer zielgerichteten CAR NK-Zell-Therapie zur Behandlung der Akuten Myeloischen Leukämie (AML)
Prof. Dr. med E. Ullrich,
Labor zelluläre Ummunologie,
Universitätsklinikum Frankfurt am Main
Klinikum für Kinder- und Jugendmedizin, Theodor-Stein-Kai 7, 60590
Frankfurt am Main
Natürliche Killer (NK) - Zellen zählen zu den Lymphozyten und sind Teil
des angeborenen Immunsystems. Sie sind in der Lage entartete, ehemals
körpereigene Zellen zu erkennen und zu töten. Aufgrund dieser
Eigenschaft kommt NK-Zellen klinisch zur Behandlung von Leukämien eine
wichtige Bedeutung zu.
In mehreren klinischen Studien wurde bereits der Einsatz von NK-Zellen
bei Patienten mit Akuter Myeloischer Leukämie (AML) getestet, und zeigte
vielversprechende Ergebnisse, ohne schwerwiegende Nebenwirkungen
hervorzurufen. Allerdings haben die bisherigen Therapie-Studien auch
gezeigt, dass NK-Zellen nur eine begrenzte Proliferationsrate (Zellneubildung)
und Überlebensdauer im Patienten haben.
In diesem Projekt soll nicht nur die Persistenz (das Fortbestehen einer Erkrankung, insbesondere das Überdauern von Krankheitserregern in Rückzugsräumen des Wirtsorganismus) von NK-Zellen durch Einsatz von Zytokinen (Proteine, die das Wachstum und die Differenzierung von Zellen regulieren), sondern auch die Zielspezifität und Zytotoxizität (die Fähigkeit chemischer Substanzen, Zellen und Gewebe zu schädigen (Toxizität). Diese Schädigung kann im Zuge einer Immunreaktion auch durch Zellen des Immunsystems vermittelt werden, z. B. durch zytotoxische T-Zellen, natürliche Killerzellen) der NK-Zellen durch Einführung von chimären Antigenrezeptoren (CARs) zum gerichteten Angriff gegen Leukämiezellen verbessert werden
Die Expression von CARs auf Effektorzellen erlaubt die direkte Erkennung Tumorassoziierter Antigene auf der Oberfläche der Tumorzellen. Nach Bindung des spezifischen Antigens vermitteln diese chimären Rezeptoren die Aktivierung der Effektorzellen, die anschließend zur Lyse der Zielzelle führt. In diesem Projekt soll die Entwicklung von CAR-exprimierenden NK-Zellen erfolgen, die als spezifische allogene Effektorzellen appliziert werden können und den Weg zur personalisierten Therapie der AML weiter voranbringen.
nach oben zur Projektseite home17/10
Unterstützung des Deutschen Registers für Stammzell-transplantationen DRST
Das Deutsche Register für Stammzelltransplantationen (DRST), gegründet am 3.4.1998 in Frankfurt am Main, ist zuständig für die zentrale Erfassung und standardisierte Auswertung aller in deutschen Trans-plantationszentren nach dem 01.01.1998 durchgeführten Transplantationen und liefert zeitnah wichtige Daten über durchgeführte Transplantationen bei verschiedenen Indikationen.
Damit stehen den Transplantationszentren wichtige
Referenzgrößen zur Beurteilung, wie auch zur Planung weiterer
Aktivitäten und Studien zur Verfügung. Darüber hinaus unterstützt das
DRST die Durchführung von nationalen und internationalen
wissenschaftlichen Studien aktiv.
Es arbeitet eng mit dem europäischen Zentrum (European Group for Blood
and Marrow Transplantation, Maastricht, Niederlande) zusammen. Dort
werden europaweit alle Blutstammzelltransplantationen in einer Datenbank
gespeichert
nach oben
zur Projektseite
home
geförderte Projekte 2018
18/1
Förderung einer internationalen wissenschaftlichen Tagung vom 25. - 27. 09. 2018 in Ffm.
Prof. Dr.
Christian Brandts, Universitätsklinik Frankfurt am Main, Direktor UCT
Universitäres Centrum für Tumorerkrankungen
Die internationale Konferenz steht unter dem Motto „Turning molecular
information into novel
cancer therapies“ und deckt alle laufenden Forschungsaktivitäten des UCT
und des DKTK (Deutsches Konsortium für Translationale Krebsforschung)
Frankfurt/Mainz ab.
Im Deutschen Konsortium für Translationale Krebsforschung kooperieren Forscher und Ärzte an acht Standorten in Deutschland, um erfolgversprechende Ansätze der Krebsforschung schneller in die klinische Praxis zu bringen.
nach oben zur Projektseite home18/2
Analyse genetische Veränderungen in linienspezifischen hämatologischen Zellen und mesenchymalen Stromazellen von Patienten mit myelodysplastischem Syndrom (MDS)
Prof. Dr. med.
Wolf-Karsten Hofmann, III. Med. Klinik Universitätsmedizin Mannheim,
Hämatologie und Onkologie,
Theodor-Kutzer-Ufer 1-3, 68167 Mannheim
Für die
langfristige und planbare Durchführung von molekulargenetischen
Untersuchungen
an selektierten hämatopoetischen Zellen von Patienten mit MDS müssen
diese unmittel-
bar nach Gewinnung der Knochenmarksproben vor Ort im wissenschaftlichen
Labor auf-
gereinigt (sortiert) werden.
Die Stiftung unterstützt die Anschaffung des dazu erforderlichen
FACS-Sortiergerätes im
Wert von fast 200.000.-- € mit einem Betrag von 40.000,-- €, um die
zukünftige Bearbeitung
von hochkompetitiven Projekten im Bereich der MDS-Forschung zu
ermöglichen.

Das FACS Zellsortiergerät
nach oben zur Projektseite home
18/3
Rolle des Insulin-ähnlichen Wachstumsfaktor-Rezeptors (IGF-1R) in der Pathogenese der akuten myeloischen Leukämie
Prof. Dr. med.
Zhixiong Li,
Professor
Dr.med. Arnold Ganser,
Medizinische
Hochschule
Hannover, Carl-Neuberg-Str.1,
30625
Hannover
Die
Behandlung der akuten myeloischen Leukämie (AML) von Erwachsenen ist
unbefriedigend, da die meisten Krebs-Gene und deren Zusammenarbeit,
welche für den Entstehungsprozess der Leukämie notwendig sind, noch
nicht hinreichend erforscht sind.
Proteintyrosinkinasen (PTKs) spielen eine wichtige Rolle bei der
malignen Transformation von Zellen.
Bei vielen Krebsarten sind Rezeptor-PTKs dereguliert, und scheinen ein
akktraktives Ziel einer selektiven molekularen Krebstherapie zu sein.
Der Insulin-ähnliche Wachstumsfaktor-Rezeptor (IGF-1R) ist wichtig für
die Entwicklung, Proliferation und Differenzierung der Zelle und
gewinnt zunehmend als Vermittler karzinogener Progression an Bedeutung.
Bisher ist wenig untersucht, ob IGF1R an der Entstehung der Leukämie
beteiligt ist. Im vorstehenden
Projekt soll ein in-vivo Modell für die IGF1R-getriebene Leukämie
etabliert werden, um die molekularen Mechanismen zu verstehen, die zur
Entwicklung humaner Leukämien führen. Es soll eine effizientere
ziel-gerichtete Therapie entwickelt werden für die AML, die durch
IGF1R-Aktivierung indiziert wird.
Schließlich soll die Arbeit zur Verbesserung der Therapie der Leukämie
beitragen.
18/4
Unterstützung des Deutschen Registers für Stammzelltransplantationen DRST
Das Deutsche Register für Stammzelltransplantationen (DRST), gegründet am 3. 4.1998 in Frankfurt am Main, ist zuständig für die zentrale Erfassung und standardisierte Auswertung aller in deutschen Trans-plantationszentren nach dem 01.01.1998 durchgeführten Transplantationen und liefert zeitnah wichtige Daten über durchgeführte Transplantationen bei verschiedenen Indikationen.
Damit stehen den Transplantationszentren wichtige Referenzgrößen zur
Beurteilung, wie auch zur Planung weiterer Aktivitäten und Studien zur
Verfügung. Darüber hinaus unterstützt das DRST die Durchführung von
nationalen und internationalen wissenschaftlichen Studien aktiv.
Es arbeitet eng mit dem europäischen Zentrum (European Group for Blood
and Marrow Transplantation, Maastricht, Niederlande) zusammen.
Dort werden europaweit alle Blutstammzelltransplantationen in
einer Datenbank gespeichert
18/5
Klonalitätsanalyse in Knochenmarkzellen von Patienten mit Myelodysplastischem Syndrom (MDS) unter antiapoptotischer Therapie mit APG101
Prof. Dr. med. Wolf-Karsten Hofmann, Prof. Dr. med. Daniel Nowak, III. Med. Klinik, Universitätsmedizin Mannheim, Hämatologie und Onkologie, Theodor-Kutzer-Ufer 1-3, 68167 Mannheim
Myelodysplastische Syndrome (MDS) sind eine Gruppe heterogener maligner Knochenmarker-krankungen, die durch eine ineffektive, dysplastische Hämatopoese mit peripheren Zytopenien, und einem stark erhöhten Risiko einer Transformation in eine sekundäre akute myeloische Leukämie (AML) gekennzeichnet sind. Bei einem wesentlichen Teil der Patienten stellt die Anämie (Blutarmut) die häufigste Indikation zum Therapie-beginn dar. Eine Anämie führt vor allem bei älteren Patienten zu Ermüdung, zu erhöhter Sturzhäufigkeit mit Frakturgefahr, zu verminderter Kognition und Lebensqualität sowie zu einem verkürzten Überleben. Für die meisten Patienten mit MDS steht deshalb die Behandlung der Anämie im Vordergrund.
Mit APG101, einem
Hemmer der Apoptose, könnte sich ein neues Therapie-Element entwickeln,
welches
in Kombination mit Erythropoetin die Behandlung der Anämie beim MDS
signifikant verbessern könnte. APG101 wirkt nach bisherigen
Erkenntnissen nicht selektiv auf die Apoptose in linienspezifischen
Zellen (hier: Erythropoese). Das vorliegende Projekt hat sich deshalb
zum Ziel gesetzt, zunächst retrospektiv an Proben von Patienten, die
bereits mit APG101 im Rahmen einer klinischen Phase II Studie behandelt
worden sind, die klonale Zusammensetzung und Entwicklung von
hämatopoetischen Zellen aller Differen-zierung zu untersuchen. Damit
könnten die dabei gewonnenen Daten Ausgangspunkt für ein sogenanntes
„molekulares Monitoring“ bei einer geplanten Phase II-II Studie sein und
routinemäßig in die Überwachung der Patienten eingeschlossen werden.
Molekulare
Untersuchungen mittels next generation sequencing (NGS) in den letzten
Jahren haben gezeigt, dass MDS Zellen im Knochenmark in den meisten
Fällen mehrere erworbene molekulare Läsionen, wie Punktmutationen und
chromosomale Veränderungen (Deletionen, Uniparentale Disomie,
Translokationen) tragen.
Diese Beobachtung hat zuletzt die Fragen aufgeworfen, ob diese
multiplen Veränderungen gleichzeitig in einem dominierenden Klon
vorkommen, oder ob mehrere Klone mit z.T. unterschiedlichen molekularen
Läsionen nebeneinander existieren und welcher klonalen Evolution diese
Klone unterliegen. Dahingehend haben mehrere Arbeiten gezeigt, dass
beide Fälle für MDS zutreffen können.
Es konnte gezeigt
werden, dass die Profile der Reihenfolge des Mutationserwerbs beim MDS
nicht zufällig sind. Als sogenannte „Gründermutationen“, die eher am
Anfang der Krankheitsentstehung und der klonalen Evolution stehen, hat
man Mutationen aus der Gruppe der Spliceosom-Mutationen und der
epigenetischen Modulatoren wie z.B. SF3B1, TET2 und DNMT3A
identifiziert.
Im Vergleich dazu treten chromosomale Veränderungen und Mutationen in
transkriptions- und Wachstums-faktoren eher später in der klonalen
Hierarchie auf.
In einer zuletzt in Blood publizierten Arbeit konnte durch NGS Analysen serieller Patientenproben gezeigt werden, dass sich die klonale Zusammensetzung im Knochenmark von MDS Patienten z.T. dramatisch und dynamisch verändert, wenn Selektionsdruck durch klinische Therapien auf die subklonale Zusammen-setzung ausgeübt wird (Mossner M et al. Blood 2016). Solche Analysen geben auf molekularer Ebene Aufschluss darüber, ob pharmakologische Substanzen überhaupt wirksam im MDS sind. Ferner erlauben sie Rückschlüsse über Molekulare Mechanismen von Sensitivität (Empfindlichkeit) bzw. Resistenz gegenüber den klinisch eingesetzten Substanzen.
In Vorbereitung
auf ein möglichst zielgerichtetes wissenschaftliches Begleitprogramm für
eine zukünftige Studie beabsichtigen wir in diesem Projekt, den Einfluss
von APG101 auf die klonale Zusammensetzung
im Knochenmark von MDS Patienten zu untersuchen, wodurch folgende
Fragestellungen verfolgt werden:
1. Lässt sich
eine Wirksamkeit von APG101 im MDS nachweisen indem es eine
systematische
Veränderung der klonalen Zusammensetzung induziert?
2. Gibt es
rekurrente molekulare Profile mit denen Klone identifiziert werden
können, die
Sensibilität
für eine Therapie mit APG101 prädiktieren?
Veröffentlichung:
https://pubmed.ncbi.nlm.nih.gov/32723198/
nach oben zur Projektseite home
18/6
Identifikation phylogenetischer Evolutionsmuster bei Patienten mit Myelofibrose
PD Dr. med.
Frederik Damm, Charité, Campus Virchow, Abt. Hämatologie, Onkologie,
Tumorimmunologie, Augustenburger Platz 1, 13353 Berlin
Krebs entwickelt sich aus einer einzelnen Zelle, die erworbene
Mutationen akkumuliert. Eine angemessene medizinische Versorgung
erfordert ein gründliches Verständnis des natürlichen Krankheitsverlaufs
ein-schließlich der Identifizierung und Reihenfolge des Auftretens der
Mutationen, der Ursprungszelle und der klonalen Organisation der
Tumorzellen.
In diesem
Projekt soll die klonale Heterogenität von Patienten mit Myelofibrose
(MF) entschlüsselt, sowie durch serielle Experimente während des
Krankheitsverlaufes und bei Transformation zu einer akuten myeloischen
Leukämie die dynamische Evolution auf Einzel-Zell Ebene verfolgt werden.
Myelofibrose und andere verwandte chronische hämatologische Neoplasien
können über viele Jahre relativ stabil sein, bevor sie zu sekundärer
akuter myeloischer Leukämie (sAML) transformieren. Daher sind diese
Krankheiten ideale Modelle, um Muster der phylogenetischen Evolution
über die Zeit zu untersuchen.
Die vorgesehenen Experimente (serielle Sequenzierung von etwa 500
Einzelzellen von fünf MF-Patienten zu drei verschiedenen Zeitpunkten des
Krankheitsverlaufs) werden als Grundlage für zahlreiche Folgestudien
dienen, einschließlich der Aufnahme von Einzelzell-Transkriptomik, und
den Grundstein legen für spätere größere Projekte.
Gefördert wird die Finanzierung von Verbrauchsmaterialen für 18 Monate.
nach oben zur Projektseite home
18/7
Aufbau einer zentralen Biomaterialbank im Rahmen einer europäischen Studie zur Erhaltungstherapie mit Panobinostat nach allogener Stammzelltrans-plantation bei Patienten mit Hochrisiko-AML oder MDS
PD Dr.
Gesine Bug, Medizinische Klinik 2, Abt. Hämatologie und Onkologie
Universitätsklinikum
Frankfurt a. M., Theodor-Stern-Kai 7, 60590 Frankfurt
Die allogene Stammzelltransplantation (SZT) kann das Überleben von Patienten mit Hochrisiko-AML oder MDS aller Altersgruppen verbessern. Leider ist die Rückfallrate dieser Patienten trotz eines potenten Transplantat-gegen-Empfänger- (graft-versus-leukemia, GvL) Effektes immer noch inakzeptabel hoch, weshalb neue Ansätze nötig sind.
Zusätzlich bleibt es ein wichtiges Ziel, die gefährliche Transplantat-gegen-Empfänger- (graft-versus-host-desease, GvHD) Reaktion zu minimieren.
Eine vielversprechende Strategie kann die Verabreichung einer epigenetischen Therapie frühzeitig nach SZT sein. Zwei vorangehende Phase I/II-Studien lassen vermuten, dass die Behandlung mit dem Histondeacety-lase-Inhibitor Panobinostat nach der SZT mit einer geringeren Rezidivrate bei gleichzeitiger GvHD-Kontrolle einhergeht. Um diese Hypothese zu untersuchen, haben wir eine große europäische Studie initiiert, in der eine Erhaltungstherapie mit Panobinostat zusammen mit Spenderlymphozyteninfusionen mit dem aktuellen Standard der Spenderlymphozyteninfusion allein bei Patienten mit Hochrisiko-AML oder MDS prospektiv randomisiert verglichen werden.
Diese ETAL-4 / HOVON-145-Studie wird von drei großen AML-Studiengruppen in Deutschland, den Niederlanden, Belgien, Norwegen, der Schweiz und Polen unterstützt und soll 350 Patienten einschließen.
Sie stellt eine einmalige Gelegenheit dar, Blutproben einer ausreichend großen Anzahl an vergleichbar behandelten Patienten zu untersuchen, um statistisch valide Ergebnisse zu erzielen, die Patienten möglichst schnell zu Gute kommen sollen.
Studienbegleitend sollen deshalb Blutproben entnommen und zusammen mit restlichem Biomaterial aus der Routinebehandlung in einer Biomaterialbank gelagert werden, um für ausgewählte Projekte der Leukämie- und Transplantationsforschung genutzt werden zu können.
Dazu gehören z. B. Untersuchungen
- von Chromosomen oder Genveränderungen in Leukämiezellen,
- von im Körper im Verlauf der Behandlung noch nachweisbaren bösartigen Zellen (minimale
Resterkrankung) und
- der Verteilung und Funktion einzelner Zellpopulationen des Spenderimmunsystems nach
der allogenen SZT.
Vorgesehen sind zwei zentrale Biobank-Standorte: in Dresden für deutsche Patienten und in Amsterdam für alle anderen Patienten.
Ziel dieses auf vier Jahre angelegten Projekts ist die Etablierung der deutschen Biobank mit Studienbezug, die alle gesetzlichen Anforderungen an die ordnungsgemäße Kodierung der Proben und den Datenschutz erfüllt und somit den Austausch von Proben erleichtern kann, die für die o. g. akademischen Forschungs-arbeiten bestimmt sind.
Dazu gehört
- die Festlegung von Standardarbeitsanweisungen für die Entnahme, Durchführung und Lagerung von
Proben von hochwertigen Knochenmark- und peripheren Blutproben von Patienten, die im Rahmen der
Studie behandelt wurden,
- die Einrichtung einer zentralen Biobank für die standardisierte Sammlung, Verarbeitung und Lagerung
von Biomaterialien aller Patienten, die in einem deutschen Studienzentrum eingeschlossen sind,
- und die Bereitstellung einer Technologieinfrastruktur für die zentrale Studiendatenbank, die sich im
Universitätsklinikum Frankfurt befindet, um eine Übersicht und den Zugang zu den asservierten Proben
zu ermöglichen, wobei die Identität und Vertraulichkeit der teilnehmenden Patienten geschützt werden.
nach oben zur Projektseite home
geförderte Projekte 2019
19/1
Genomische Analyse der akuten lymphatischen Leukämie mit MLL-Fusionsgen
PD Dr. med. rer nat. Thomas Burmeister,
Laborleiter Tumorgenetik – Molekulardiagnostik
im Labor Berlin,
Universitätssmed. Berlin Charité Campus Virchow-Klinikum, Augustenburger
Platz 1, 13353 Berlin
Etwa 10 Prozent der Patienten mit akuten Leukämien (sowohl myeloische,
als auch lymphatische)
weisen Aberrationen (Abweichungen)
des MLL
(KMT2A)-Gens auf. Gehäuft finden sich MLLAberrationen
auch bei sekundären Leukämien, z. B. infolge eines Myelodysplastischen
Syndroms nach vorangegangener Chemotherapie.
Die betroffenen Patienten gelten durchgängig als Hochrisiko-Patienten,
die einer Transplantation zugeführt werden sollten.
Bisher gibt es keine vergleichbar wirksame gezielte Therapiemöglichkeit,
wie es beispielsweise bei den BCR-ABL-positiven Leukämien der
Fall ist. Die genauen Mechanismen der Leukämogenese (Entwicklung
der Leukämie)
sind bei den MLL-rearrangierten Leukämien auch noch unzureichend
verstanden.
Das vorliegende Projekt befasst sich mit der genaueren genomischen Charakterisierung der akuten lymphatische Leukämie mit MLL-Aberration (Abweichung).
Im Rahmen eines vorangegangenen Projektes wurden Komplettgenomanalysen und Transkriptomanalysen (die Summe aller zu einem best. Zeitpunkt in einer Zelle von DNA in RNA umgeschriebenen Gene, d. h. alle in einer Zelle hergestellten RNA-Moleküle) bei MLL-rearrangierten Patientenproben durchgeführt.
Insgesamt wurden drei ausgewählte Patientenproben mit MLL-rearrangierter akuter lymphatischer Leukämie Komplett-Genom sequenziert (DNA) und davon jeweils bei zwei Proben sowohl eine Leukämieprobe, als auch eine Remissionsprobe. Zusätzlich wurden bei 32 ausgewählten Patientenproben mit MLL-rearrangierter akuter lymphatischer Leukämie eine komplette Transkriptom-sequenzierung durchgeführt. Als "Referenz" dienten hier ebenfalls im Rahmen des Projektes generierte Transkriptomsequenzierungen aus hochaufge-reinigten CD19-positiven Blutzellen von vier gesunden Spendern.
Im Rahmen dieser Komplett-Genom-Analysen wurden Gene identifiziert, die rekurrent alteriert bzw. Gen-muster identifiziert, die überexprimiert bzw. reprimiert waren. (Wikipedia rekurrent – Wiederauftreten einer Krankheit oder Symptoms) alteriert=erregt, aufgeregt, aufregen) (Genexpression oder Exprimierung = die genetische Information eines Gens 'Abschntt der DNA' die zum Ausdruck kommt) Diese Gene sind hochinteressante Kandidaten für eventuelle Kofaktoren bei der Leukämogenese MLLrearrangierter Leukämien. Derartige Kofaktoren sind bisher nicht hinreichend charakterisiert.
Die Förderdauer des genannten Forschungsprojektes betrug 24 Monate. Leider war es innerhalb dieser Zeitdauer nicht möglich, die Arbeiten vollständig abzuschließen und eine Verlängerung des Projektes war nicht möglich. Mit dem vorliegenden Antrag sollen daher die notwendigen abschließenden Arbeiten finanziert werden. Die Genom- und Transkriptomsequenzierungen sind bereits erfolgt und weitestgehend ausgewertet (weitere Sub-Auswertungen können noch erfolgen).
Die jetzt noch notwendigen Schritte umfassen die Verifizierung der gefundenen rekurrenten Veränderungen an einem größeren Patientenkollektiv von MLL-rearrangierten Patientenproben mittels konventioneller PCRs und real-time quantitativer RT-PCRs, sowie einzelner Sequenzierungen.
Das Projekt verspricht, interessante Ergebnisse zu liefern, vor allem
auch deswegen, weil es sich um einen qualitativ sehr hochwertigen
Datensatz handelt. Es wurden nur sehr ausgewählte Proben in die Analyse
miteinbezogen (Blastenanteil > 80 %) und rigorose Qualitätskriterien für
den Einschluss von Patientenroben angelegt.
nach oben
zur Projektseite
home
19/2
Rolle des Axl-Rezeptors in der Pathogenese
der akuten myeloischen
Leukämie
Prof. Dr. med. Zhixiong Li,
Professor
Dr.med. Arnold Ganser, Medizinische
Hochschule Hannover,
Carl-Neuberg-Str.1,
30625 Hannover
Deregulation von
Proteintyrosinkinasen (PTKs) tritt bei vielen Krebsarten häufig auf und
stellt ein attraktives Ziel einer selektiven molekularen Krebstherapie
dar. Das Axl-Gen (auch als UFO, ARK und TYRO7 bekannt) wurde vor zwei
Jahrzehnten bei zwei Patienten mit chronischer myeloischer Leukämie
entdeckt. Axl wurde später als RTK der TAMUnterfamilie (TYRO3, Axl und
MER) klassifiziert. Es gibt zunehmend Hinweise darauf, dass die
Überexpression oder Überaktivierung des Axl-Proteins mit der Förderung
multipler tumori-gener Prozesse korreliert.
Ein hohes Maß an Axl-Expression ist mit einer schlechten Prognose bei
verschiedenen Krebsarten wie Osteosarkom, Brust- und Lungenkrebs sowie
AML (Akute myeloische Leukämie) assoziiert.
In humanen akuten
Leukämien ist bisher nicht gut untersucht, ob und wie Axl an der
Entstehung der Leukämie beteiligt ist. Insbesondere gibt es noch keine
Modelle von Axl-getriebener Leukämie.
In einer Studie haben wir Phosphorylierung von Axl in 33% Patienten mit
AML beobachtet.
Daher möchten wir hier Modelle für Axl-getriebene Leukämie etablieren, die molekularen Mechanismen verstehen und eine effizientere molekulare Therapie entwickeln.
Die beschriebenen Arbeiten sollen helfen, die molekulare Therapie von AML zu verbessern
nach oben zur Projektseite home19/3
Genetische Analyse von Knochenmark-Stromazellen von Pytienten mit myelo-dysplastischem Syndrom (MDS) auf Einzel-Zell-Niveau
Prof. Dr. med. Wolf-Karsten Hofmann, Dr. med.
Johann-Christoph Jann,
III.
Med. Klinik
Universitätsmedizin Mannheim, Hämatologie und Onkologie,
Theodor-Kutzer-Ufer 1-3,
68167 Mannheim
Die Untersuchung von genetischen Veränderungen hat in den letzten Jahren
sehr stark dazu beigetragen, dass bösartige Bluterkrankungen immer
besser diagnostiziert und inzwischen mit verschiedenen Medika-menten
erfolgreich behandelt werden können. Um diese Erfolge weiter ausbauen zu
können, ist es zunehmend erforderlich, die Ursachen der Erkrankung –
die in mehr als 90 % der Fälle nicht bekannt sind –
zu untersuchen.
Die Anwendung der Techniken des DNA-Sequenzierens war bisher auf die Analyse von sogenannten „Zellgruppen“ (also mehrere tausend Zellen aus dem Blut oder dem Knochenmark) beschränkt. Das hatte insbesondere technische Gründe (Menge des zur Verfügung stehenden Zellmaterials). Sehr moderne und aktuelle Entwicklungen auf dem Gebiet der genomischen Sequenzierung erlauben es nun, einzelne Zellen des menschlichen Körpers vollständig genetisch zu untersuchen und damit ein zellspezifisches molekulares Profil zu erstellen. Diese Technik (Single-Cell NGS, Einzel-Zell-Next-Generation-Sequencing) wird die molekulare Diagnostik und den Erkenntnisgewinn über Krankheiten der Blutbildung revolutionieren, da es damit möglich sein wird, einzelne krankheitsspezifische (Tumor-) Zellen genau zu charakterisieren. Bei hämatologischen Erkrankungen könnte so ein neuer Meilenstein bei der Untersuchung der Pathophysiologie und Mechanismen dieser bösartigen Veränderungen entstehen.
Im
vorliegenden Projekt soll diese innovative und bahnbrechende Technik auf
die Analyse von einzelnen Knochenmarkzellen von Patienten mit
Myelodysplastischem Syndrom (MDS) angewandt werden, um weitere
Erkenntnisse über die zu Grunde liegenden Ursachen der Erkrankung zu
gewinnen.
nach oben
zur Projektseite
home
19/4
Unterstützung des Deutschen Registers für Stammzelltransplantationen
DRST
Das Deutsche Register für Stammzelltransplantationen (DRST), gegründet am 3. 4.1998 in Frankfurt am Main, ist zuständig für die zentrale Erfassung und standardisierte Auswertung aller in deutschen Trans-plantationszentren nach dem 01.01.1998 durchgeführten Transplantationen und liefert zeitnah wichtige Daten über durchgeführte Transplantationen bei verschiedenen Indikationen.
Damit stehen den Transplantationszentren wichtige Referenzgrößen zur
Beurteilung, wie auch zur Planung weiterer Aktivitäten und Studien zur
Verfügung. Darüber hinaus unterstützt das DRST die Durchführung von
nationalen und internationalen wissenschaftlichen Studien aktiv.
Es arbeitet eng mit dem europäischen Zentrum (European Group for Blood
and Marrow Transplantation, Maastricht, Niederlande) zusammen.
Dort werden europaweit alle Blutstammzelltransplantationen in
einer Datenbank gespeichert
nach oben zur Projektseite home
19/5
Pre- und intratherapeutische
zelluläre Überwachung zur Anpassung zyto-toxischer Behandlungen bei
aggressiven Lymphomen
Prof. Dr. med. P. Markus Deckert, Dr. Julia Schröder. Med. Hochschule
Brandenburg Theodor Fontane, Städt. Klinikum Brandenburg, Klinik für
Hämatologie, Onkologie und Palliativmedizin,
Hochstraße 29,
14770 Brandenburg
Ungeachtet moderner Entwicklungen molekular gerichteter Medikamente
bleibt die zytostatische Therapie (Chemotherapie) ein
Grundpfeiler der Krebstherapie.
Im Gegensatz zu molekular gerichteten Therapien bestehen jedoch kaum
etablierte Methoden, ihre Wirksamkeit und Verträglichkeit individuell
vorherzusagen und die Dosis entsprechend zu bestimmen.
in diesem Projekt sollen biologische Parameter der Zellschädigung (DNA
Doppelstrang-brüche und Telomerlänge), sowie mögliche prognostische
Parameter (CD30, MYC,BCL2 und BCL6) auf ihre Aussagekraft für das
Ansprechen und die Toxizität der Therapie aggressiver Lymphome
untersucht werden.
Bisher überwiegend in Gewebeproben untersucht, sollen diese Parameter
analog dem Konzept der "Flüssigbiopbsie" (liquid biopsy)
(diagnostischer Nachweis von Tumorzellen bzw. Tumor-DNA im Blut) an
Lymphozyten vergleichend zu Lymphomgewebe angewandt werden.
Darüber hinaus wurden verschiedene microRNAs (miRNAs) als pathogene
Faktoren und potenzielle Biomarker für aggressive Lymphome beschrieben,
deren Bedeutung für Diagnose und Therapie bisher noch unklar sind.
Ein methodisch unterschiedlicher Arm mit gleicher Zielsetzug soll daher
den Vorhersage-wert ausgewählter miRNAs aus dem peripheren Blut
evaluieren
nach oben
zur Projektseite
home